{"title":"通过小基因剪接分析揭示剪接变异对遗传性听力障碍的功能影响。","authors":"Lara Emily Rosso, Giulia Pianigiani, Anna Morgan, Elisa Rubinato, Elisa Paccagnella, Stefania Lenarduzzi, Anita Wischmeijer, Beatrice Spedicati, Giorgia Girotto","doi":"10.3390/biomedicines13092245","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background/Objectives</b>: Hereditary hearing loss (HHL) is a genetically heterogeneous condition, involving more than 150 genes in non-syndromic cases and associated with over 400 distinct disorders in syndromic forms. Although whole-exome sequencing (WES) has markedly increased diagnostic yield, a substantial number of cases remain unsolved, often due to intronic variants that affect splicing and are difficult to interpret. This study aimed to characterize the potential impact of intronic variants predicted to alter splicing in families affected by HHL. <b>Methods</b>: The effect of seven intronic variants, previously identified in a diagnostic setting by WES within <i>ADGRV1</i>, <i>ATP11A</i>, <i>GSDME</i>, <i>OTOF</i>, <i>OTOGL</i>, and <i>USH2A</i> genes, was evaluated. To functionally validate these predictions, in vitro minigene splicing assays were subsequently performed. <b>Results</b>: All the identified variants were predicted to disrupt normal RNA splicing. The functional studies with minigene assays confirmed this observation and showed that the tested variants induced both exon skipping and activation of cryptic splice sites. In five out of seven cases, these splicing alterations caused a frameshift and introduced a premature termination codon, ultimately resulting in nonsense-mediated mRNA decay and protein degradation. <b>Conclusions</b>: This study expands the mutational spectrum of HL-related genes and highlights the importance of integrating in silico predictions with minigene assays. Such a combined approach is crucial for accurate interpretation of splicing variants, particularly when patient-derived RNA samples are unavailable.</p>","PeriodicalId":8937,"journal":{"name":"Biomedicines","volume":"13 9","pages":""},"PeriodicalIF":3.9000,"publicationDate":"2025-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12467345/pdf/","citationCount":"0","resultStr":"{\"title\":\"Unraveling the Functional Impact of Splicing Variants in Inherited Hearing Disorders Through Minigene Splicing Assays.\",\"authors\":\"Lara Emily Rosso, Giulia Pianigiani, Anna Morgan, Elisa Rubinato, Elisa Paccagnella, Stefania Lenarduzzi, Anita Wischmeijer, Beatrice Spedicati, Giorgia Girotto\",\"doi\":\"10.3390/biomedicines13092245\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><b>Background/Objectives</b>: Hereditary hearing loss (HHL) is a genetically heterogeneous condition, involving more than 150 genes in non-syndromic cases and associated with over 400 distinct disorders in syndromic forms. Although whole-exome sequencing (WES) has markedly increased diagnostic yield, a substantial number of cases remain unsolved, often due to intronic variants that affect splicing and are difficult to interpret. This study aimed to characterize the potential impact of intronic variants predicted to alter splicing in families affected by HHL. <b>Methods</b>: The effect of seven intronic variants, previously identified in a diagnostic setting by WES within <i>ADGRV1</i>, <i>ATP11A</i>, <i>GSDME</i>, <i>OTOF</i>, <i>OTOGL</i>, and <i>USH2A</i> genes, was evaluated. To functionally validate these predictions, in vitro minigene splicing assays were subsequently performed. <b>Results</b>: All the identified variants were predicted to disrupt normal RNA splicing. The functional studies with minigene assays confirmed this observation and showed that the tested variants induced both exon skipping and activation of cryptic splice sites. In five out of seven cases, these splicing alterations caused a frameshift and introduced a premature termination codon, ultimately resulting in nonsense-mediated mRNA decay and protein degradation. <b>Conclusions</b>: This study expands the mutational spectrum of HL-related genes and highlights the importance of integrating in silico predictions with minigene assays. Such a combined approach is crucial for accurate interpretation of splicing variants, particularly when patient-derived RNA samples are unavailable.</p>\",\"PeriodicalId\":8937,\"journal\":{\"name\":\"Biomedicines\",\"volume\":\"13 9\",\"pages\":\"\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2025-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12467345/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomedicines\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://doi.org/10.3390/biomedicines13092245\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomedicines","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.3390/biomedicines13092245","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Unraveling the Functional Impact of Splicing Variants in Inherited Hearing Disorders Through Minigene Splicing Assays.
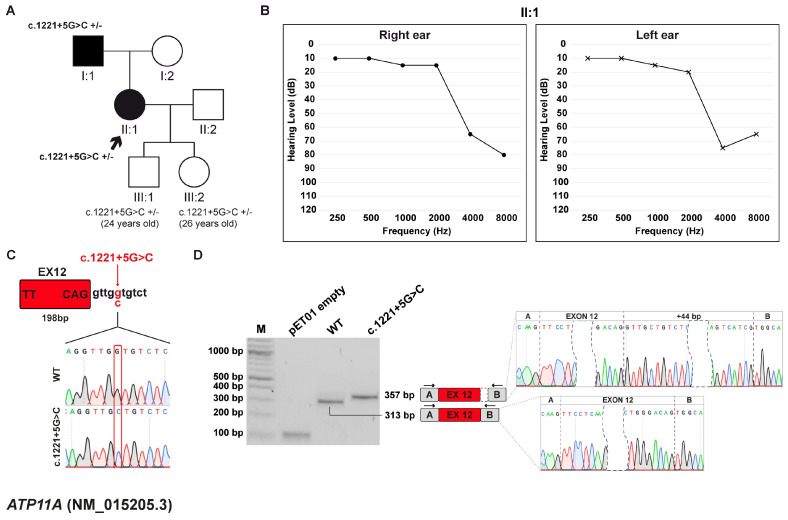
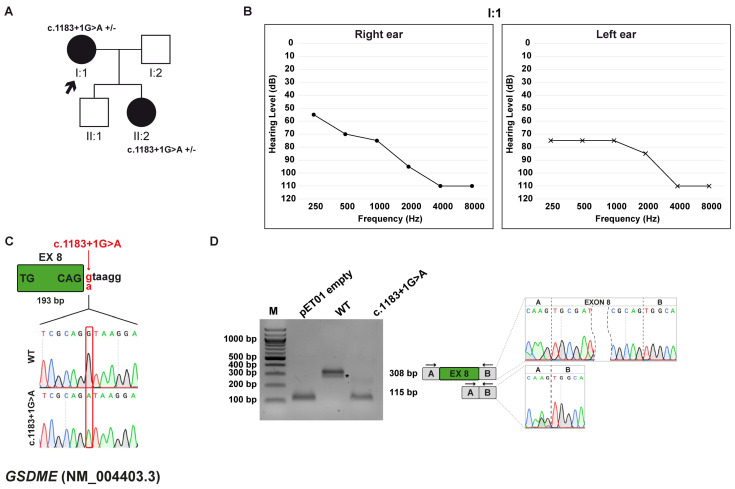
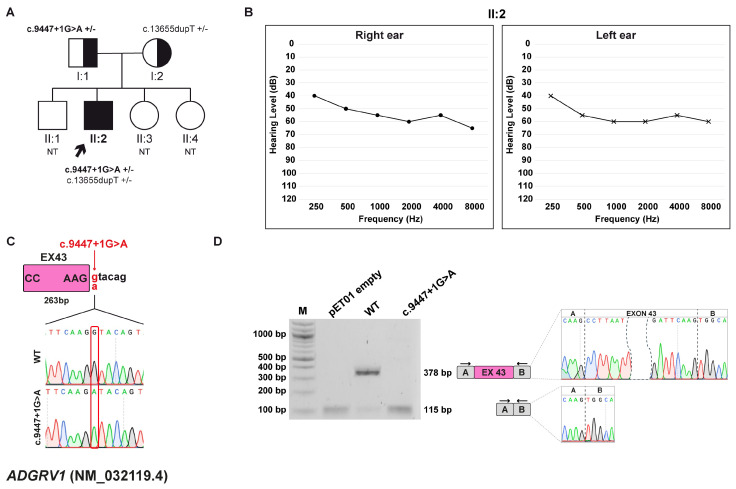
Background/Objectives: Hereditary hearing loss (HHL) is a genetically heterogeneous condition, involving more than 150 genes in non-syndromic cases and associated with over 400 distinct disorders in syndromic forms. Although whole-exome sequencing (WES) has markedly increased diagnostic yield, a substantial number of cases remain unsolved, often due to intronic variants that affect splicing and are difficult to interpret. This study aimed to characterize the potential impact of intronic variants predicted to alter splicing in families affected by HHL. Methods: The effect of seven intronic variants, previously identified in a diagnostic setting by WES within ADGRV1, ATP11A, GSDME, OTOF, OTOGL, and USH2A genes, was evaluated. To functionally validate these predictions, in vitro minigene splicing assays were subsequently performed. Results: All the identified variants were predicted to disrupt normal RNA splicing. The functional studies with minigene assays confirmed this observation and showed that the tested variants induced both exon skipping and activation of cryptic splice sites. In five out of seven cases, these splicing alterations caused a frameshift and introduced a premature termination codon, ultimately resulting in nonsense-mediated mRNA decay and protein degradation. Conclusions: This study expands the mutational spectrum of HL-related genes and highlights the importance of integrating in silico predictions with minigene assays. Such a combined approach is crucial for accurate interpretation of splicing variants, particularly when patient-derived RNA samples are unavailable.
BiomedicinesBiochemistry, Genetics and Molecular Biology-General Biochemistry,Genetics and Molecular Biology
CiteScore
5.20
自引率
8.50%
发文量
2823
审稿时长
8 weeks
期刊介绍:
Biomedicines (ISSN 2227-9059; CODEN: BIOMID) is an international, scientific, open access journal on biomedicines published quarterly online by MDPI.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


