Pavel Tiuleanu, Ivan V. Ivanov, Kirill V. Rychev, Natalia E. Grammatikova, Irina P. Andreeva, Vitaly G. Grigorenko, Alexander M. Scherbakov, Alexey M. Egorov, Andrey E. Shchekotikhin
{"title":"2-偶氮吡啶的合成及其作为金属β-内酰胺酶抑制剂的潜力评价","authors":"Pavel Tiuleanu, Ivan V. Ivanov, Kirill V. Rychev, Natalia E. Grammatikova, Irina P. Andreeva, Vitaly G. Grigorenko, Alexander M. Scherbakov, Alexey M. Egorov, Andrey E. Shchekotikhin","doi":"10.1007/s00044-025-03436-y","DOIUrl":null,"url":null,"abstract":"<div><p>Bacterial resistance to β-lactam antibiotics has emerged as a major challenge in healthcare. This form of antibiotic resistance is driven by the ability of pathogens to produce β-lactamases, which are divided into four classes (A-D) according to the Ambler classification. The most concerning are metallo-β-lactamases (MBLs) of class B, with the New Delhi Metallo-β-lactamase (NDM) enzyme family being among the most clinically significant. These zinc-dependent enzymes can inactivate almost all β-lactam antibiotics, and, to date, no effective inhibitors for this type of β-lactamase have been developed. Certain derivatives of indole-2-carboxylic acid and azoles have been shown to inhibit New Delhi metallo-β-lactamase-1 (NDM-1) by coordinating with Zn<sup>2+</sup> ions and specifically interacting with key amino acid residues in the active site of the enzyme. However, the antibacterial potential of azolylindoles as metallo-β-lactamase inhibitors remains unexplored. In searches of novel scaffolds for the development of metallo-β-lactamase inhibitors a strategy for modifying a known NDM-1 inhibitor chemotype based on indole-2-carboxylic acid is proposed. This approach leads to the synthesis of previously unreported 2-azolylindoles incorporating triazole, thiadiazole, oxadiazole, tetrazole, and tetrazolylmethyl moieties. Synthetic methodologies for the preparation of intermediates and target compounds were optimized and adapted. Obtained compounds have demonstrated the ability to inhibit NDM-1 across a broad concentration range (IC<sub>50</sub> = 40 nM–15 µM), highlighting the significant influence of the azole nuclei structure on in the enzyme inhibition. The docking-predicted binding poses of the most active compounds in active site of NDM-1 closely matched with the experimental ligand orientation and revealed interactions with key amino acid residues and Zn<sup>2+</sup> ions. For the most potent lead-compounds, the effect on the activity of meropenem and cefepime against NDM-1-producing strains of <i>E. coli</i> and <i>K. pneumoniae</i> was evaluated, as these antibiotics are highly relevant in antimicrobial therapy. Cytotoxicity of the synthesized series was evaluated using the noncancerous HaCaT keratinocyte cell line. The most active NDM-1 inhibitors, including the paternal acid <b>1</b> and the tetrazole analogue <b>19</b>, demonstrated low cytotoxicity (IC<sub>50</sub> > 50 µM), supporting their potential as safe candidates. In contrast, compounds containing triazole, thiadiazole, or oxadiazole moieties showed increased cytotoxicity, which also limits the further development of compounds <b>25</b> and <b>26</b> as NDM-1 inhibitors. Thus, among all synthesized compounds, only 2-tetrazolylindole derivative was identified as a potential candidate for further development of novel MBL inhibitors.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":699,"journal":{"name":"Medicinal Chemistry Research","volume":"34 8","pages":"1714 - 1732"},"PeriodicalIF":3.1000,"publicationDate":"2025-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"2-azolylindoles: synthesis and evaluation of their potential as metallo-β-lactamase inhibitors\",\"authors\":\"Pavel Tiuleanu, Ivan V. Ivanov, Kirill V. Rychev, Natalia E. Grammatikova, Irina P. Andreeva, Vitaly G. Grigorenko, Alexander M. Scherbakov, Alexey M. Egorov, Andrey E. Shchekotikhin\",\"doi\":\"10.1007/s00044-025-03436-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Bacterial resistance to β-lactam antibiotics has emerged as a major challenge in healthcare. This form of antibiotic resistance is driven by the ability of pathogens to produce β-lactamases, which are divided into four classes (A-D) according to the Ambler classification. The most concerning are metallo-β-lactamases (MBLs) of class B, with the New Delhi Metallo-β-lactamase (NDM) enzyme family being among the most clinically significant. These zinc-dependent enzymes can inactivate almost all β-lactam antibiotics, and, to date, no effective inhibitors for this type of β-lactamase have been developed. Certain derivatives of indole-2-carboxylic acid and azoles have been shown to inhibit New Delhi metallo-β-lactamase-1 (NDM-1) by coordinating with Zn<sup>2+</sup> ions and specifically interacting with key amino acid residues in the active site of the enzyme. However, the antibacterial potential of azolylindoles as metallo-β-lactamase inhibitors remains unexplored. In searches of novel scaffolds for the development of metallo-β-lactamase inhibitors a strategy for modifying a known NDM-1 inhibitor chemotype based on indole-2-carboxylic acid is proposed. This approach leads to the synthesis of previously unreported 2-azolylindoles incorporating triazole, thiadiazole, oxadiazole, tetrazole, and tetrazolylmethyl moieties. Synthetic methodologies for the preparation of intermediates and target compounds were optimized and adapted. Obtained compounds have demonstrated the ability to inhibit NDM-1 across a broad concentration range (IC<sub>50</sub> = 40 nM–15 µM), highlighting the significant influence of the azole nuclei structure on in the enzyme inhibition. The docking-predicted binding poses of the most active compounds in active site of NDM-1 closely matched with the experimental ligand orientation and revealed interactions with key amino acid residues and Zn<sup>2+</sup> ions. For the most potent lead-compounds, the effect on the activity of meropenem and cefepime against NDM-1-producing strains of <i>E. coli</i> and <i>K. pneumoniae</i> was evaluated, as these antibiotics are highly relevant in antimicrobial therapy. Cytotoxicity of the synthesized series was evaluated using the noncancerous HaCaT keratinocyte cell line. The most active NDM-1 inhibitors, including the paternal acid <b>1</b> and the tetrazole analogue <b>19</b>, demonstrated low cytotoxicity (IC<sub>50</sub> > 50 µM), supporting their potential as safe candidates. In contrast, compounds containing triazole, thiadiazole, or oxadiazole moieties showed increased cytotoxicity, which also limits the further development of compounds <b>25</b> and <b>26</b> as NDM-1 inhibitors. Thus, among all synthesized compounds, only 2-tetrazolylindole derivative was identified as a potential candidate for further development of novel MBL inhibitors.</p><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":699,\"journal\":{\"name\":\"Medicinal Chemistry Research\",\"volume\":\"34 8\",\"pages\":\"1714 - 1732\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2025-07-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Medicinal Chemistry Research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00044-025-03436-y\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Medicinal Chemistry Research","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00044-025-03436-y","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
2-azolylindoles: synthesis and evaluation of their potential as metallo-β-lactamase inhibitors
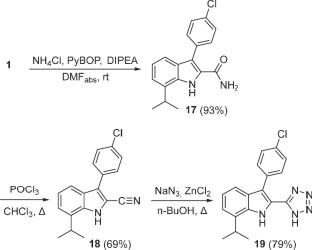
Bacterial resistance to β-lactam antibiotics has emerged as a major challenge in healthcare. This form of antibiotic resistance is driven by the ability of pathogens to produce β-lactamases, which are divided into four classes (A-D) according to the Ambler classification. The most concerning are metallo-β-lactamases (MBLs) of class B, with the New Delhi Metallo-β-lactamase (NDM) enzyme family being among the most clinically significant. These zinc-dependent enzymes can inactivate almost all β-lactam antibiotics, and, to date, no effective inhibitors for this type of β-lactamase have been developed. Certain derivatives of indole-2-carboxylic acid and azoles have been shown to inhibit New Delhi metallo-β-lactamase-1 (NDM-1) by coordinating with Zn2+ ions and specifically interacting with key amino acid residues in the active site of the enzyme. However, the antibacterial potential of azolylindoles as metallo-β-lactamase inhibitors remains unexplored. In searches of novel scaffolds for the development of metallo-β-lactamase inhibitors a strategy for modifying a known NDM-1 inhibitor chemotype based on indole-2-carboxylic acid is proposed. This approach leads to the synthesis of previously unreported 2-azolylindoles incorporating triazole, thiadiazole, oxadiazole, tetrazole, and tetrazolylmethyl moieties. Synthetic methodologies for the preparation of intermediates and target compounds were optimized and adapted. Obtained compounds have demonstrated the ability to inhibit NDM-1 across a broad concentration range (IC50 = 40 nM–15 µM), highlighting the significant influence of the azole nuclei structure on in the enzyme inhibition. The docking-predicted binding poses of the most active compounds in active site of NDM-1 closely matched with the experimental ligand orientation and revealed interactions with key amino acid residues and Zn2+ ions. For the most potent lead-compounds, the effect on the activity of meropenem and cefepime against NDM-1-producing strains of E. coli and K. pneumoniae was evaluated, as these antibiotics are highly relevant in antimicrobial therapy. Cytotoxicity of the synthesized series was evaluated using the noncancerous HaCaT keratinocyte cell line. The most active NDM-1 inhibitors, including the paternal acid 1 and the tetrazole analogue 19, demonstrated low cytotoxicity (IC50 > 50 µM), supporting their potential as safe candidates. In contrast, compounds containing triazole, thiadiazole, or oxadiazole moieties showed increased cytotoxicity, which also limits the further development of compounds 25 and 26 as NDM-1 inhibitors. Thus, among all synthesized compounds, only 2-tetrazolylindole derivative was identified as a potential candidate for further development of novel MBL inhibitors.
期刊介绍:
Medicinal Chemistry Research (MCRE) publishes papers on a wide range of topics, favoring research with significant, new, and up-to-date information. Although the journal has a demanding peer review process, MCRE still boasts rapid publication, due in part, to the length of the submissions. The journal publishes significant research on various topics, many of which emphasize the structure-activity relationships of molecular biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


