R. Sayyadi Kordabadi, S. A. S. Hashemi, O. Alizadeh
{"title":"马拉维克衍生物治疗HIV的计算研究:QSAR和分子对接方法","authors":"R. Sayyadi Kordabadi, S. A. S. Hashemi, O. Alizadeh","doi":"10.1134/S1990793125700198","DOIUrl":null,"url":null,"abstract":"<p>This research utilized four different methods to investigate the structure-activity relationships of 25 derivatives of Maraviroc. The combination of Genetic Algorithms-Artificial Neural Networks (GA–ANN) and Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) demonstrated superior performance among both linear and nonlinear methods. The study identified specific descriptors, such as atomic van der Waals volumes, polarizability, and atomic masses, as significant in the Genetic Algorithms–Artificial Neural Networks (GA-ANN) method for biological activity assessment. In terms of lipophilicity, descriptors related to Verhaar Algae base-line toxicity and polarizability were highlighted in the Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) method. Molecular docking analysis revealed that Maraviroc derivative number 22 with 5UIW receptor exhibited the lowest affinity but the highest number of hydrogen bonds. The Monte Carlo technique, utilizing CORAL software, pinpointed essential molecular characteristics linked to both biological activity (–logIC<sub>50</sub>) and lipophilicity (XLOGP). These features encompassed the existence of cyclic rings with branching, <i>sp</i><sup>2</sup> carbon linked to a ring, exclusive presence of double bonds, Nitrogen attachment to cyclic rings, Nitrogen presence in double bonds, Fluorine atom connection to branching, presence of branching, and Nitrogen atom linkage to a ring. The research found that the combined use of GA-ANN, MLR-ICA, Monte Carlo method, and molecular docking can enhance understanding of the relationship between physico-chemical descriptors and drug mechanisms, aiding in the design of new drugs.</p>","PeriodicalId":768,"journal":{"name":"Russian Journal of Physical Chemistry B","volume":"19 2","pages":"348 - 361"},"PeriodicalIF":1.4000,"publicationDate":"2025-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational Studies on Maraviroc Derivatives for HIV Treatment: QSAR and Molecular Docking Approaches\",\"authors\":\"R. Sayyadi Kordabadi, S. A. S. Hashemi, O. Alizadeh\",\"doi\":\"10.1134/S1990793125700198\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>This research utilized four different methods to investigate the structure-activity relationships of 25 derivatives of Maraviroc. The combination of Genetic Algorithms-Artificial Neural Networks (GA–ANN) and Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) demonstrated superior performance among both linear and nonlinear methods. The study identified specific descriptors, such as atomic van der Waals volumes, polarizability, and atomic masses, as significant in the Genetic Algorithms–Artificial Neural Networks (GA-ANN) method for biological activity assessment. In terms of lipophilicity, descriptors related to Verhaar Algae base-line toxicity and polarizability were highlighted in the Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) method. Molecular docking analysis revealed that Maraviroc derivative number 22 with 5UIW receptor exhibited the lowest affinity but the highest number of hydrogen bonds. The Monte Carlo technique, utilizing CORAL software, pinpointed essential molecular characteristics linked to both biological activity (–logIC<sub>50</sub>) and lipophilicity (XLOGP). These features encompassed the existence of cyclic rings with branching, <i>sp</i><sup>2</sup> carbon linked to a ring, exclusive presence of double bonds, Nitrogen attachment to cyclic rings, Nitrogen presence in double bonds, Fluorine atom connection to branching, presence of branching, and Nitrogen atom linkage to a ring. The research found that the combined use of GA-ANN, MLR-ICA, Monte Carlo method, and molecular docking can enhance understanding of the relationship between physico-chemical descriptors and drug mechanisms, aiding in the design of new drugs.</p>\",\"PeriodicalId\":768,\"journal\":{\"name\":\"Russian Journal of Physical Chemistry B\",\"volume\":\"19 2\",\"pages\":\"348 - 361\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2025-07-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Russian Journal of Physical Chemistry B\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S1990793125700198\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"PHYSICS, ATOMIC, MOLECULAR & CHEMICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of Physical Chemistry B","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S1990793125700198","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"PHYSICS, ATOMIC, MOLECULAR & CHEMICAL","Score":null,"Total":0}
Computational Studies on Maraviroc Derivatives for HIV Treatment: QSAR and Molecular Docking Approaches
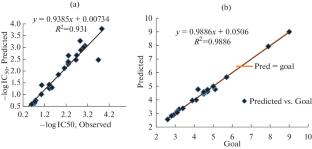
This research utilized four different methods to investigate the structure-activity relationships of 25 derivatives of Maraviroc. The combination of Genetic Algorithms-Artificial Neural Networks (GA–ANN) and Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) demonstrated superior performance among both linear and nonlinear methods. The study identified specific descriptors, such as atomic van der Waals volumes, polarizability, and atomic masses, as significant in the Genetic Algorithms–Artificial Neural Networks (GA-ANN) method for biological activity assessment. In terms of lipophilicity, descriptors related to Verhaar Algae base-line toxicity and polarizability were highlighted in the Multiple Linear Regression-Imperialist Competitive Algorithm (MLR-ICA) method. Molecular docking analysis revealed that Maraviroc derivative number 22 with 5UIW receptor exhibited the lowest affinity but the highest number of hydrogen bonds. The Monte Carlo technique, utilizing CORAL software, pinpointed essential molecular characteristics linked to both biological activity (–logIC50) and lipophilicity (XLOGP). These features encompassed the existence of cyclic rings with branching, sp2 carbon linked to a ring, exclusive presence of double bonds, Nitrogen attachment to cyclic rings, Nitrogen presence in double bonds, Fluorine atom connection to branching, presence of branching, and Nitrogen atom linkage to a ring. The research found that the combined use of GA-ANN, MLR-ICA, Monte Carlo method, and molecular docking can enhance understanding of the relationship between physico-chemical descriptors and drug mechanisms, aiding in the design of new drugs.
期刊介绍:
Russian Journal of Physical Chemistry B: Focus on Physics is a journal that publishes studies in the following areas: elementary physical and chemical processes; structure of chemical compounds, reactivity, effect of external field and environment on chemical transformations; molecular dynamics and molecular organization; dynamics and kinetics of photoand radiation-induced processes; mechanism of chemical reactions in gas and condensed phases and at interfaces; chain and thermal processes of ignition, combustion and detonation in gases, two-phase and condensed systems; shock waves; new physical methods of examining chemical reactions; and biological processes in chemical physics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


