晶体碎片筛选揭示了PYCR1抑制剂设计的新起点
IF 4.7
2区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
吡咯-5-羧酸(P5C)还原酶催化脯氨酸生物合成的最后一步。人类P5C还原酶异构体1 (PYCR1)通过其在氧化还原稳态、胶原生成和脯氨酸-P5C循环中的作用,已成为支持癌症进展的关键代谢酶。尽管PYCR1作为治疗靶点具有相关性,但抑制PYCR1的结构和化学努力仍然有限,并且主要集中在脯氨酸类似物上。在这里,我们报告了针对PYCR1的第一个晶体片段筛选(XFS)运动,使用96个化合物的化学多样性文库。我们解决了12个共晶结构,其中配体占据了P5C和NADH的结合口袋,包括跨越两个区域的双位点配体。在新发现的部分中,磺胺和磺胺酸基团作为PYCR1活性位点羧酸基团的显着等构替代出现。一些化合物的芳香取代基在烟酰胺结合位点附近发现了一个隐蔽的亚袋。有趣的是,通常存在于已知PYCR1抑制剂中的卤素取代芳香环表现出不同的结合方向,反映了结合子口袋中相互作用的灵活性和多样性。高分辨率结构揭示了配体诱导的PYCR1构象变化,其中一些涉及重大重排。分子动力学模拟表明,这些构象在无配体的酶中是可获得的,这强调了PYCR1活性位点的内在可塑性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
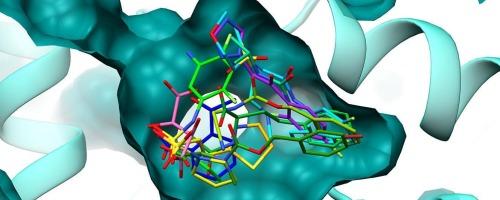
Crystallographic fragment screening reveals new starting points for PYCR1 inhibitor design
Pyrroline-5-carboxylate (P5C) reductase catalyzes the final step in proline biosynthesis. Human P5C reductase isoform 1 (PYCR1) has emerged as a key metabolic enzyme supporting cancer progression through its roles in redox homeostasis, collagen production, and the proline-P5C cycle. Despite its relevance as a therapeutic target, structural and chemical efforts to inhibit PYCR1 remain limited and have largely focused on proline analogs. Here, we report the first crystallographic fragment screening (XFS) campaign against PYCR1, employing a chemically diverse library of 96 compounds. We solved twelve co-crystal structures, featuring ligands occupying the P5C and NADH binding pockets, including dual-site ligands that span both regions. Among the newly identified moieties, sulfonamide and sulfamate groups emerged as notable isosteric replacements for the carboxylate group in the PYCR1 active site. Aromatic substituents in several compounds revealed a cryptic subpocket near the nicotinamide-binding site. Interestingly, halogen-substituted aromatic rings, often present in known PYCR1 inhibitors, exhibited distinct binding orientations, reflecting the flexibility and diversity of interactions in the binding subpockets. High-resolution structures revealed ligand-induced conformational changes in PYCR1, some involving significant rearrangements. Molecular dynamics simulations indicated that these conformations are accessible in the ligand-free enzyme, underscoring the intrinsic plasticity of PYCR1's active site.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Bioorganic Chemistry
生物-生化与分子生物学
CiteScore
9.70
自引率
3.90%
发文量
679
审稿时长
31 days
期刊介绍:
Bioorganic Chemistry publishes research that addresses biological questions at the molecular level, using organic chemistry and principles of physical organic chemistry. The scope of the journal covers a range of topics at the organic chemistry-biology interface, including: enzyme catalysis, biotransformation and enzyme inhibition; nucleic acids chemistry; medicinal chemistry; natural product chemistry, natural product synthesis and natural product biosynthesis; antimicrobial agents; lipid and peptide chemistry; biophysical chemistry; biological probes; bio-orthogonal chemistry and biomimetic chemistry.
For manuscripts dealing with synthetic bioactive compounds, the Journal requires that the molecular target of the compounds described must be known, and must be demonstrated experimentally in the manuscript. For studies involving natural products, if the molecular target is unknown, some data beyond simple cell-based toxicity studies to provide insight into the mechanism of action is required. Studies supported by molecular docking are welcome, but must be supported by experimental data. The Journal does not consider manuscripts that are purely theoretical or computational in nature.
The Journal publishes regular articles, short communications and reviews. Reviews are normally invited by Editors or Editorial Board members. Authors of unsolicited reviews should first contact an Editor or Editorial Board member to determine whether the proposed article is within the scope of the Journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


