{"title":"基于多模态自定义门控制的蛋白质-配体结合力鲁棒预测。","authors":"Bofei Xu, Wenting Tang, Danial Muhammad, Yuqi Yin, Zhirong Liu, Zhaoxi Sun","doi":"10.1021/acs.jcim.5c01668","DOIUrl":null,"url":null,"abstract":"<p><p>The main protease (Mpro) is a critical target in the design of antiviral drugs against coronaviruses, while accurately predicting the binding affinity between small molecules and this target remains a key challenge. In the recent Polaris challenge of blind drug-potency prediction targeting SARS-CoV-2 and MERS-CoV Mpro, we developed a multimodal multitask graph attention network based on the customized gate control framework (abbreviated as MultiMolCGC). Our team achieved top performance among all participating teams in the blind prediction challenge. In this paper, we detail the model development and further explorations in terms of pretraining, adjusting the model architecture, and many others. Our model consistently outperforms traditional machine learning baselines, demonstrating the effectiveness of end-to-end deep learning in capturing complex molecular interactions. Integrating multimodal representations proved essential, and the multitask specialized gating architecture outperformed both single-task and nonspecialized multitask variants, highlighting the value of tailored knowledge sharing. While auxiliary loss weighting and hyperparameter tuning offered modest improvements, incorporating predicted structural data unexpectedly reduced performance, likely due to structural uncertainty. Notably, pretraining on large-scale synthetic docking data sets significantly enhanced performance in low-data scenarios, reducing dependence on experimental pIC<sub>50</sub> data. The numerical results highlight the potential of MultiMolCGC as a robust and accurate deep-learning framework for protein-ligand binding in future studies.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2025-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Robust Prediction of Protein-Ligand Binding Potency with Multi-modal Customized Gate Control.\",\"authors\":\"Bofei Xu, Wenting Tang, Danial Muhammad, Yuqi Yin, Zhirong Liu, Zhaoxi Sun\",\"doi\":\"10.1021/acs.jcim.5c01668\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The main protease (Mpro) is a critical target in the design of antiviral drugs against coronaviruses, while accurately predicting the binding affinity between small molecules and this target remains a key challenge. In the recent Polaris challenge of blind drug-potency prediction targeting SARS-CoV-2 and MERS-CoV Mpro, we developed a multimodal multitask graph attention network based on the customized gate control framework (abbreviated as MultiMolCGC). Our team achieved top performance among all participating teams in the blind prediction challenge. In this paper, we detail the model development and further explorations in terms of pretraining, adjusting the model architecture, and many others. Our model consistently outperforms traditional machine learning baselines, demonstrating the effectiveness of end-to-end deep learning in capturing complex molecular interactions. Integrating multimodal representations proved essential, and the multitask specialized gating architecture outperformed both single-task and nonspecialized multitask variants, highlighting the value of tailored knowledge sharing. While auxiliary loss weighting and hyperparameter tuning offered modest improvements, incorporating predicted structural data unexpectedly reduced performance, likely due to structural uncertainty. Notably, pretraining on large-scale synthetic docking data sets significantly enhanced performance in low-data scenarios, reducing dependence on experimental pIC<sub>50</sub> data. The numerical results highlight the potential of MultiMolCGC as a robust and accurate deep-learning framework for protein-ligand binding in future studies.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.5c01668\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.5c01668","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Robust Prediction of Protein-Ligand Binding Potency with Multi-modal Customized Gate Control.
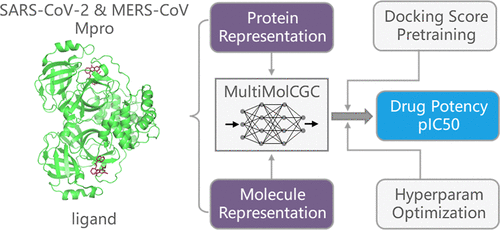
The main protease (Mpro) is a critical target in the design of antiviral drugs against coronaviruses, while accurately predicting the binding affinity between small molecules and this target remains a key challenge. In the recent Polaris challenge of blind drug-potency prediction targeting SARS-CoV-2 and MERS-CoV Mpro, we developed a multimodal multitask graph attention network based on the customized gate control framework (abbreviated as MultiMolCGC). Our team achieved top performance among all participating teams in the blind prediction challenge. In this paper, we detail the model development and further explorations in terms of pretraining, adjusting the model architecture, and many others. Our model consistently outperforms traditional machine learning baselines, demonstrating the effectiveness of end-to-end deep learning in capturing complex molecular interactions. Integrating multimodal representations proved essential, and the multitask specialized gating architecture outperformed both single-task and nonspecialized multitask variants, highlighting the value of tailored knowledge sharing. While auxiliary loss weighting and hyperparameter tuning offered modest improvements, incorporating predicted structural data unexpectedly reduced performance, likely due to structural uncertainty. Notably, pretraining on large-scale synthetic docking data sets significantly enhanced performance in low-data scenarios, reducing dependence on experimental pIC50 data. The numerical results highlight the potential of MultiMolCGC as a robust and accurate deep-learning framework for protein-ligand binding in future studies.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


