André de Freitas Gonçalves, , , Emerson Parazzi Lyra, , , Sayali Ramdas Chavan, , , Philip L. Llewellyn, , , Luis Fernando Mercier Franco, , and , Yann Magnin*,
{"title":"CO2在亚纳米多孔材料中吸附和扩散的基础:CALF-20的应用","authors":"André de Freitas Gonçalves, , , Emerson Parazzi Lyra, , , Sayali Ramdas Chavan, , , Philip L. Llewellyn, , , Luis Fernando Mercier Franco, , and , Yann Magnin*, ","doi":"10.1021/acs.jpcc.5c03130","DOIUrl":null,"url":null,"abstract":"<p >We propose an approach for approximating the thermodynamics and kinetics of guest molecules in nanoporous materials. This statistical mechanics-based method requires a minimal set of parameters as input and has been applied to CO<sub>2</sub> molecules in the recently highlighted CALF-20 metal–organic framework for adsorption at different temperatures. The physical parameters of the adsorption model are extracted from one CO<sub>2</sub> isotherm at a given temperature and analyzed by the adsorption energy distribution method. The model is then used to approximate isotherms at different temperatures, Henry’s constant, saturation density, as well as enthalpies of adsorption at infinite dilution. The approach was further applied in MFI zeolite with CH<sub>4</sub> guests to ensure the transferability of the method. We then express molecular kinetics through the transition state theory, allowing one to approximate molecular diffusion, in part from thermodynamics, and further compare self-diffusion coefficients with molecular dynamics used as a numerical experiment. The approach proposed allows to express the molecular adsorption and self-diffusion in CALF-20 based on a formalism fed by physical parameters. The model proposed may be used to choose an appropriate isotherm model or, alternatively, can serve to help in setting an initial guess in a standard fitting procedure.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 40","pages":"18190–18199"},"PeriodicalIF":3.2000,"publicationDate":"2025-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fundamental of CO2 Adsorption and Diffusion in Subnanoporous Materials: Application to CALF-20\",\"authors\":\"André de Freitas Gonçalves, , , Emerson Parazzi Lyra, , , Sayali Ramdas Chavan, , , Philip L. Llewellyn, , , Luis Fernando Mercier Franco, , and , Yann Magnin*, \",\"doi\":\"10.1021/acs.jpcc.5c03130\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We propose an approach for approximating the thermodynamics and kinetics of guest molecules in nanoporous materials. This statistical mechanics-based method requires a minimal set of parameters as input and has been applied to CO<sub>2</sub> molecules in the recently highlighted CALF-20 metal–organic framework for adsorption at different temperatures. The physical parameters of the adsorption model are extracted from one CO<sub>2</sub> isotherm at a given temperature and analyzed by the adsorption energy distribution method. The model is then used to approximate isotherms at different temperatures, Henry’s constant, saturation density, as well as enthalpies of adsorption at infinite dilution. The approach was further applied in MFI zeolite with CH<sub>4</sub> guests to ensure the transferability of the method. We then express molecular kinetics through the transition state theory, allowing one to approximate molecular diffusion, in part from thermodynamics, and further compare self-diffusion coefficients with molecular dynamics used as a numerical experiment. The approach proposed allows to express the molecular adsorption and self-diffusion in CALF-20 based on a formalism fed by physical parameters. The model proposed may be used to choose an appropriate isotherm model or, alternatively, can serve to help in setting an initial guess in a standard fitting procedure.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 40\",\"pages\":\"18190–18199\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03130\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03130","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Fundamental of CO2 Adsorption and Diffusion in Subnanoporous Materials: Application to CALF-20
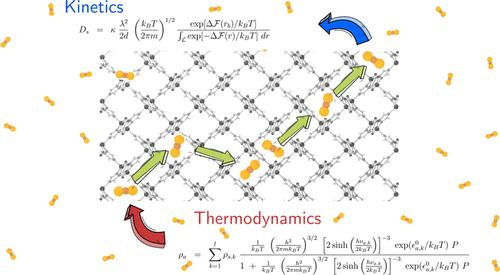
We propose an approach for approximating the thermodynamics and kinetics of guest molecules in nanoporous materials. This statistical mechanics-based method requires a minimal set of parameters as input and has been applied to CO2 molecules in the recently highlighted CALF-20 metal–organic framework for adsorption at different temperatures. The physical parameters of the adsorption model are extracted from one CO2 isotherm at a given temperature and analyzed by the adsorption energy distribution method. The model is then used to approximate isotherms at different temperatures, Henry’s constant, saturation density, as well as enthalpies of adsorption at infinite dilution. The approach was further applied in MFI zeolite with CH4 guests to ensure the transferability of the method. We then express molecular kinetics through the transition state theory, allowing one to approximate molecular diffusion, in part from thermodynamics, and further compare self-diffusion coefficients with molecular dynamics used as a numerical experiment. The approach proposed allows to express the molecular adsorption and self-diffusion in CALF-20 based on a formalism fed by physical parameters. The model proposed may be used to choose an appropriate isotherm model or, alternatively, can serve to help in setting an initial guess in a standard fitting procedure.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


