Garrett M Janzen, Blake T Inderski, Jennifer Chang, Zebulun W Arendsee, Alicia Janas-Martindale, Mia Kim Torchetti, Amy L Baker, Tavis K Anderson
{"title":"2009年至2022年美国猪甲型流感病毒基因组多样性的来源和汇","authors":"Garrett M Janzen, Blake T Inderski, Jennifer Chang, Zebulun W Arendsee, Alicia Janas-Martindale, Mia Kim Torchetti, Amy L Baker, Tavis K Anderson","doi":"10.1128/jvi.00541-25","DOIUrl":null,"url":null,"abstract":"<p><p>Influenza A virus (IAV) in swine in the U.S. is surveilled to monitor genetic evolution to inform intervention efforts and aid pandemic preparedness. We describe data from the U.S. Department of Agriculture National Surveillance Plan for Influenza A Virus in Pigs from 2009 to 2022. Clinical respiratory cases were subtyped, followed by sequencing of hemagglutinin (HA) and neuraminidase (NA), and a subset of viruses was whole genome sequenced. Phylogenetic analysis identified geographic and temporal IAV reassortment hotspots. Regions acting as IAV genomic diversity sources or sinks were quantified, and dissemination was qualified and modeled. The dominant IAV clades were H1N2 (1B.2.1), H3N2 (1990.4.a), and H1N1 (H1-1A.3.3.3-c3). Internal genes were classified as triple-reassortant (T) or pandemic 2009 (P), and three genome constellations represented 73.5% of detections across the last 2 years. In some years, the distribution of IAV diversity was so narrow that it presented a statistical signal associated with local adaptation. We also demonstrated that the source of most IAV genomic diversity was in Midwest states (IL, MO, IA), and while this was correlated with swine inventory, the emergence and persistence of diversity were tied to swine transport across the U.S. The continued regional detection of unique HA, NA, and genome constellations provides support for targeted interventions to improve animal health and enhance pandemic preparedness.IMPORTANCEVariation in the genetic diversity of influenza A virus (IAV) in swine through time and between regions impacts control efforts. This study quantified the genomic diversity of swine IAV collected from 2009 to 2022 at regional and national levels and modeled sources and sinks of that diversity. Seasonal patterns of IAV transmission were observed, and some locations contributed disproportionately to the emergence of genomic diversity. Minor groups of viruses had the potential to disseminate across the U.S. with animal movement. The identification of these patterns demonstrates the importance of a robust surveillance system to inform vaccine updates that reflect regional patterns of genetic diversity. We show how preemptive interventions in swine IAV diversity hubs could reduce reassortment and the emergence of novel genomic diversity, and how these efforts are likely to reduce the transmission of IAV within swine and between swine and humans.</p>","PeriodicalId":17583,"journal":{"name":"Journal of Virology","volume":"99 9","pages":"e0054125"},"PeriodicalIF":3.8000,"publicationDate":"2025-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12455956/pdf/","citationCount":"0","resultStr":"{\"title\":\"Sources and sinks of influenza A virus genomic diversity in swine from 2009 to 2022 in the United States.\",\"authors\":\"Garrett M Janzen, Blake T Inderski, Jennifer Chang, Zebulun W Arendsee, Alicia Janas-Martindale, Mia Kim Torchetti, Amy L Baker, Tavis K Anderson\",\"doi\":\"10.1128/jvi.00541-25\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Influenza A virus (IAV) in swine in the U.S. is surveilled to monitor genetic evolution to inform intervention efforts and aid pandemic preparedness. We describe data from the U.S. Department of Agriculture National Surveillance Plan for Influenza A Virus in Pigs from 2009 to 2022. Clinical respiratory cases were subtyped, followed by sequencing of hemagglutinin (HA) and neuraminidase (NA), and a subset of viruses was whole genome sequenced. Phylogenetic analysis identified geographic and temporal IAV reassortment hotspots. Regions acting as IAV genomic diversity sources or sinks were quantified, and dissemination was qualified and modeled. The dominant IAV clades were H1N2 (1B.2.1), H3N2 (1990.4.a), and H1N1 (H1-1A.3.3.3-c3). Internal genes were classified as triple-reassortant (T) or pandemic 2009 (P), and three genome constellations represented 73.5% of detections across the last 2 years. In some years, the distribution of IAV diversity was so narrow that it presented a statistical signal associated with local adaptation. We also demonstrated that the source of most IAV genomic diversity was in Midwest states (IL, MO, IA), and while this was correlated with swine inventory, the emergence and persistence of diversity were tied to swine transport across the U.S. The continued regional detection of unique HA, NA, and genome constellations provides support for targeted interventions to improve animal health and enhance pandemic preparedness.IMPORTANCEVariation in the genetic diversity of influenza A virus (IAV) in swine through time and between regions impacts control efforts. This study quantified the genomic diversity of swine IAV collected from 2009 to 2022 at regional and national levels and modeled sources and sinks of that diversity. Seasonal patterns of IAV transmission were observed, and some locations contributed disproportionately to the emergence of genomic diversity. Minor groups of viruses had the potential to disseminate across the U.S. with animal movement. The identification of these patterns demonstrates the importance of a robust surveillance system to inform vaccine updates that reflect regional patterns of genetic diversity. We show how preemptive interventions in swine IAV diversity hubs could reduce reassortment and the emergence of novel genomic diversity, and how these efforts are likely to reduce the transmission of IAV within swine and between swine and humans.</p>\",\"PeriodicalId\":17583,\"journal\":{\"name\":\"Journal of Virology\",\"volume\":\"99 9\",\"pages\":\"e0054125\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2025-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12455956/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Virology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1128/jvi.00541-25\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Virology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1128/jvi.00541-25","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/26 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"VIROLOGY","Score":null,"Total":0}
Sources and sinks of influenza A virus genomic diversity in swine from 2009 to 2022 in the United States.
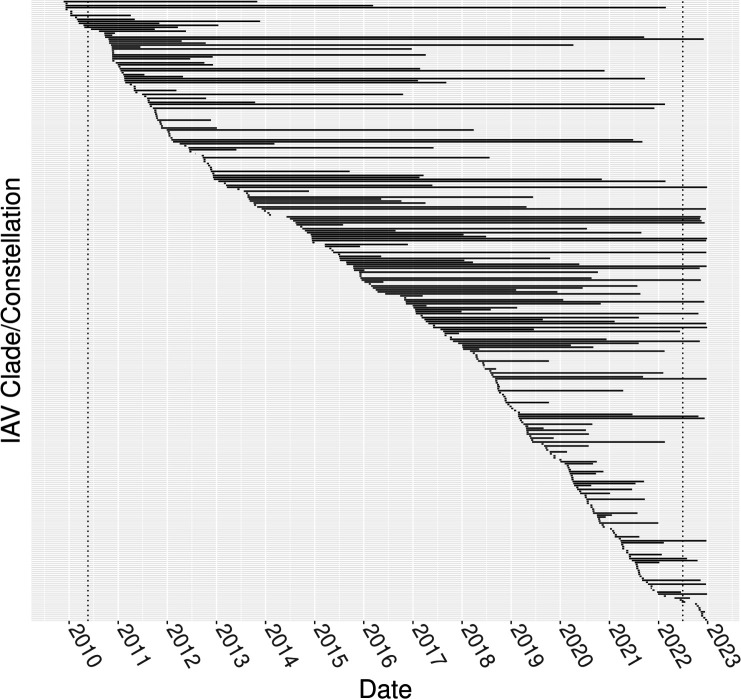
Influenza A virus (IAV) in swine in the U.S. is surveilled to monitor genetic evolution to inform intervention efforts and aid pandemic preparedness. We describe data from the U.S. Department of Agriculture National Surveillance Plan for Influenza A Virus in Pigs from 2009 to 2022. Clinical respiratory cases were subtyped, followed by sequencing of hemagglutinin (HA) and neuraminidase (NA), and a subset of viruses was whole genome sequenced. Phylogenetic analysis identified geographic and temporal IAV reassortment hotspots. Regions acting as IAV genomic diversity sources or sinks were quantified, and dissemination was qualified and modeled. The dominant IAV clades were H1N2 (1B.2.1), H3N2 (1990.4.a), and H1N1 (H1-1A.3.3.3-c3). Internal genes were classified as triple-reassortant (T) or pandemic 2009 (P), and three genome constellations represented 73.5% of detections across the last 2 years. In some years, the distribution of IAV diversity was so narrow that it presented a statistical signal associated with local adaptation. We also demonstrated that the source of most IAV genomic diversity was in Midwest states (IL, MO, IA), and while this was correlated with swine inventory, the emergence and persistence of diversity were tied to swine transport across the U.S. The continued regional detection of unique HA, NA, and genome constellations provides support for targeted interventions to improve animal health and enhance pandemic preparedness.IMPORTANCEVariation in the genetic diversity of influenza A virus (IAV) in swine through time and between regions impacts control efforts. This study quantified the genomic diversity of swine IAV collected from 2009 to 2022 at regional and national levels and modeled sources and sinks of that diversity. Seasonal patterns of IAV transmission were observed, and some locations contributed disproportionately to the emergence of genomic diversity. Minor groups of viruses had the potential to disseminate across the U.S. with animal movement. The identification of these patterns demonstrates the importance of a robust surveillance system to inform vaccine updates that reflect regional patterns of genetic diversity. We show how preemptive interventions in swine IAV diversity hubs could reduce reassortment and the emergence of novel genomic diversity, and how these efforts are likely to reduce the transmission of IAV within swine and between swine and humans.
期刊介绍:
Journal of Virology (JVI) explores the nature of the viruses of animals, archaea, bacteria, fungi, plants, and protozoa. We welcome papers on virion structure and assembly, viral genome replication and regulation of gene expression, genetic diversity and evolution, virus-cell interactions, cellular responses to infection, transformation and oncogenesis, gene delivery, viral pathogenesis and immunity, and vaccines and antiviral agents.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


