非甾体抗炎药偶联超短拟肽作为抗甲氧西林耐药金黄色葡萄球菌的有效抗菌药物
IF 4.7
2区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
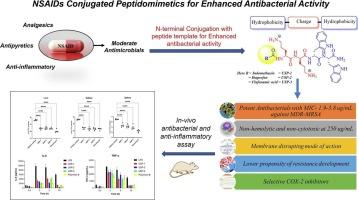
为了设计针对多药耐药(MDR)病原体的新疗法,我们在这里报道了一系列药物偶联的超短肽模拟物。鉴于非甾体抗炎药(NSAIDs)具有中等抑菌活性,本研究旨在通过与两亲性四肽模板(H-Orn-Orn-Trp-Trp-NH₂)偶联来增强其抑菌潜力,同时保持其抗炎作用。采用固相多肽法合成了这些NSAID-peptide缀合物,并对其抗菌性能进行了评价。在文库中,3种拟肽物(USP-1、USP-2、USP-3)对革兰氏阳性和革兰氏阴性细菌(包括耐多药甲氧西林金黄色葡萄球菌)均表现出较强的抑菌活性,最低抑菌浓度(mic)在1.9 ~ 62.5 μg/mL之间。有趣的是,这些强效化合物显示出非溶血性和非细胞毒性,其浓度比其抗菌mic高100倍。抗菌模式的作用研究表明,这些拟肽主要是去极化和破坏细菌细胞膜的mic。此外,即使在各自的亚mic浓度持续暴露18次传代后,MRSA也未观察到耐药性的发展。重要的是,缀合的非甾体抗炎药通过选择性抑制COX-2酶保留了其抗炎活性。最后,在小鼠体内的功效研究表明,优化的偶联物在单次腹腔剂量为10 mg/kg时,可显著降低细菌负荷和炎症。总的来说,这些偶联物具有抗菌和抗炎双重特性,加上耐药倾向低和良好的安全性,代表它们是治疗耐药感染的有希望的候选者。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Non-steroidal anti-inflammatory drugs conjugated ultra-short Peptidomimetics as potent Antibacterials against methicillin resistant S. aureus with anti-inflammatory activity
Towards design of novel therapeutics against multidrug-resistant (MDR) pathogens, we reported here a focused series of drug-conjugated ultrashort peptidomimetics. Recognizing the moderate antibacterial activity of non-steroidal anti-inflammatory drugs (NSAIDs), the aim of this study was to enhance their antibacterial potential by conjugating them with an amphiphilic tetrapeptide template (H-Orn-Orn-Trp-Trp-NH₂), while preserving their anti-inflammatory effects. These NSAID-peptide conjugates were synthesized via solid phase peptide synthesis and their antibacterial properties were assessed. Within the library, three peptidomimetics (USP-1, USP-2, USP-3) showed potent antibacterial activity, with minimum inhibitory concentrations (MICs) ranging from 1.9 to 62.5 μg/mL against both Gram-positive and Gram-negative bacteria, including MDR methicillin resistant S. aureus. Interestingly, these potent compounds demonstrated non-hemolytic and non-cytotoxic with up to >100-fold higher concentrations than their antibacterial MICs. The antibacterial mode of action studies revealed that these peptidomimetics primarily depolarized and disrupted the bacterial cell membranes at their MICs. Furthermore, resistance development was not observed in MRSA even after 18 passages with continue exposure of their respective sub-MIC concentrations. Importantly, the conjugated NSAIDs retained their anti-inflammatory activity, by selectively inhibiting COX-2 enzyme. Finally, in vivo efficacy studies in mice revealed that optimized conjugates significantly reduced bacterial load and inflammation at a single intra-peritoneal dose of 10 mg/kg. Overall, these conjugates exhibit dual antibacterial and anti-inflammatory properties, coupled with a low propensity for resistance and favourable safety profile, representing them as promising candidates for the treatment of drug-resistant infections.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Bioorganic Chemistry
生物-生化与分子生物学
CiteScore
9.70
自引率
3.90%
发文量
679
审稿时长
31 days
期刊介绍:
Bioorganic Chemistry publishes research that addresses biological questions at the molecular level, using organic chemistry and principles of physical organic chemistry. The scope of the journal covers a range of topics at the organic chemistry-biology interface, including: enzyme catalysis, biotransformation and enzyme inhibition; nucleic acids chemistry; medicinal chemistry; natural product chemistry, natural product synthesis and natural product biosynthesis; antimicrobial agents; lipid and peptide chemistry; biophysical chemistry; biological probes; bio-orthogonal chemistry and biomimetic chemistry.
For manuscripts dealing with synthetic bioactive compounds, the Journal requires that the molecular target of the compounds described must be known, and must be demonstrated experimentally in the manuscript. For studies involving natural products, if the molecular target is unknown, some data beyond simple cell-based toxicity studies to provide insight into the mechanism of action is required. Studies supported by molecular docking are welcome, but must be supported by experimental data. The Journal does not consider manuscripts that are purely theoretical or computational in nature.
The Journal publishes regular articles, short communications and reviews. Reviews are normally invited by Editors or Editorial Board members. Authors of unsolicited reviews should first contact an Editor or Editorial Board member to determine whether the proposed article is within the scope of the Journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


