{"title":"线粒体乙酰乙酰辅酶a硫酶缺乏症:新生儿筛查新发现3例,证实C4OH升高的意义","authors":"Alessandra Vasco, Clarissa Berardo, Simona Lucchi, Laura Cappelletti, Giulio Tamburello, Salvatore Fazzone, Alessia Mauri, Francesca Fiumani, Diana Postorivo, Luisella Alberti, Michela Perrone Donnorso, Serena Gasperini, Francesca Furlan, Laura Fiori, Stephana Carelli, Laura Assunta Saielli, Cristina Montrasio, Cristina Cereda","doi":"10.3390/ijns11030076","DOIUrl":null,"url":null,"abstract":"<p><p>Acetoacetyl-CoA thiolase deficiency, also known as Beta-ketothiolase deficiency (BKTD), is an autosomal recessive organic aciduria included in the Italian newborn screening (NBS) panel. It is caused by mutations in the <i>ACAT1</i> gene, which encodes the mitochondrial acetyl-CoA acetyltransferase. Its deficiency impairs the degradation of isoleucine and acetoacetyl-CoA, leading to the accumulation of toxic metabolites. We describe three cases of BKTD. The first newborn showed increase in C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. Urinary organic acids (uOAs) revealed marked excretion of 2-methyl-3-hydroxybutyrate. Tiglylglycine was absent. Genetic testing identified the compound heterozygosity for two pathogenic <i>ACAT1</i> variants. The second patient showed increased levels of C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. uOAs revealed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous VUS in <i>ACAT1</i> was identified. The third case showed elevation of C4DC/C5OH, C3DC/C4OH in the NBS, with a slight increase in C5:1. uOAs showed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous missense VUS was identified in the <i>ACAT1</i> gene. BKTD exhibited variable NBS biochemical phenotypes across the three cases. While C5OH and C5:1, the primary markers, were not consistently elevated in all our cases, C4OH strongly increased in all three. Our findings support the use of C4OH in a combined marker strategy to improve BKTD NBS.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 3","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12452439/pdf/","citationCount":"0","resultStr":"{\"title\":\"Mitochondrial Acetoacetyl-CoA Thiolase Deficiency: Three New Cases Detected by Newborn Screening Confirming the Significance of C4OH Elevation.\",\"authors\":\"Alessandra Vasco, Clarissa Berardo, Simona Lucchi, Laura Cappelletti, Giulio Tamburello, Salvatore Fazzone, Alessia Mauri, Francesca Fiumani, Diana Postorivo, Luisella Alberti, Michela Perrone Donnorso, Serena Gasperini, Francesca Furlan, Laura Fiori, Stephana Carelli, Laura Assunta Saielli, Cristina Montrasio, Cristina Cereda\",\"doi\":\"10.3390/ijns11030076\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Acetoacetyl-CoA thiolase deficiency, also known as Beta-ketothiolase deficiency (BKTD), is an autosomal recessive organic aciduria included in the Italian newborn screening (NBS) panel. It is caused by mutations in the <i>ACAT1</i> gene, which encodes the mitochondrial acetyl-CoA acetyltransferase. Its deficiency impairs the degradation of isoleucine and acetoacetyl-CoA, leading to the accumulation of toxic metabolites. We describe three cases of BKTD. The first newborn showed increase in C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. Urinary organic acids (uOAs) revealed marked excretion of 2-methyl-3-hydroxybutyrate. Tiglylglycine was absent. Genetic testing identified the compound heterozygosity for two pathogenic <i>ACAT1</i> variants. The second patient showed increased levels of C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. uOAs revealed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous VUS in <i>ACAT1</i> was identified. The third case showed elevation of C4DC/C5OH, C3DC/C4OH in the NBS, with a slight increase in C5:1. uOAs showed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous missense VUS was identified in the <i>ACAT1</i> gene. BKTD exhibited variable NBS biochemical phenotypes across the three cases. While C5OH and C5:1, the primary markers, were not consistently elevated in all our cases, C4OH strongly increased in all three. Our findings support the use of C4OH in a combined marker strategy to improve BKTD NBS.</p>\",\"PeriodicalId\":14159,\"journal\":{\"name\":\"International Journal of Neonatal Screening\",\"volume\":\"11 3\",\"pages\":\"\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2025-09-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12452439/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Neonatal Screening\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/ijns11030076\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11030076","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Mitochondrial Acetoacetyl-CoA Thiolase Deficiency: Three New Cases Detected by Newborn Screening Confirming the Significance of C4OH Elevation.
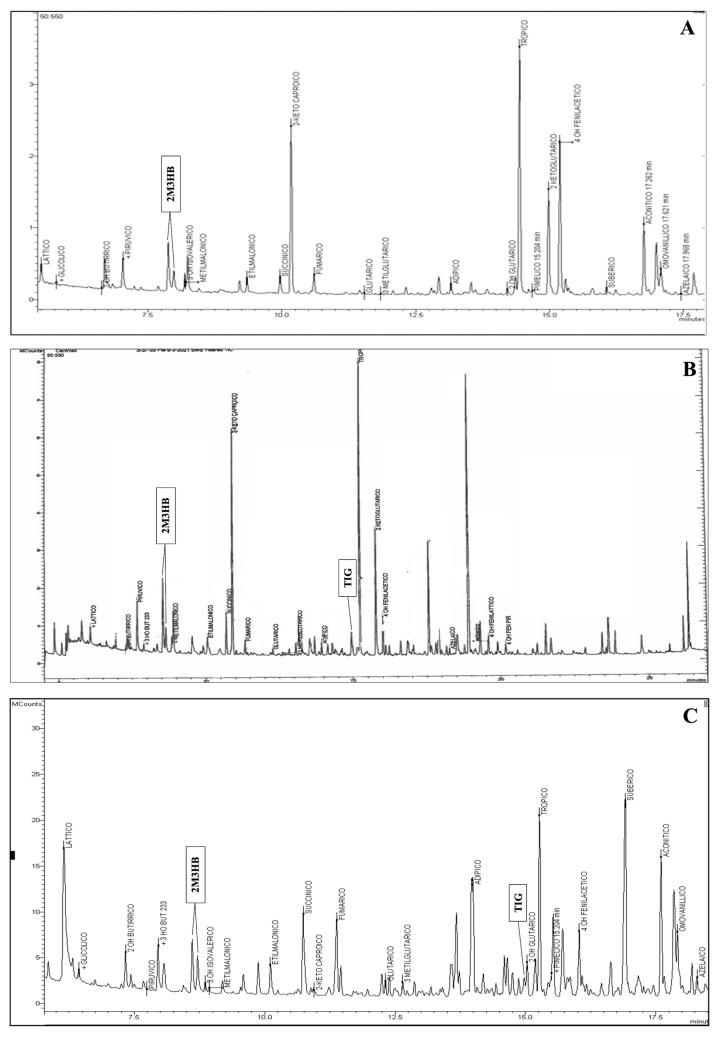
Acetoacetyl-CoA thiolase deficiency, also known as Beta-ketothiolase deficiency (BKTD), is an autosomal recessive organic aciduria included in the Italian newborn screening (NBS) panel. It is caused by mutations in the ACAT1 gene, which encodes the mitochondrial acetyl-CoA acetyltransferase. Its deficiency impairs the degradation of isoleucine and acetoacetyl-CoA, leading to the accumulation of toxic metabolites. We describe three cases of BKTD. The first newborn showed increase in C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. Urinary organic acids (uOAs) revealed marked excretion of 2-methyl-3-hydroxybutyrate. Tiglylglycine was absent. Genetic testing identified the compound heterozygosity for two pathogenic ACAT1 variants. The second patient showed increased levels of C5:1, C4DC/C5OH, C3DC/C4OH in the NBS. uOAs revealed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous VUS in ACAT1 was identified. The third case showed elevation of C4DC/C5OH, C3DC/C4OH in the NBS, with a slight increase in C5:1. uOAs showed 2-methyl-3-hydroxybutyrate and tiglylglycine. A homozygous missense VUS was identified in the ACAT1 gene. BKTD exhibited variable NBS biochemical phenotypes across the three cases. While C5OH and C5:1, the primary markers, were not consistently elevated in all our cases, C4OH strongly increased in all three. Our findings support the use of C4OH in a combined marker strategy to improve BKTD NBS.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


