一种新型查尔酮衍生物的合成、表征和生物学评价:光谱、DFT、对接、ADMET和MD研究
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
以2-乙酰吩噻嗪和2-羟基-3-甲氧基苯甲醛为原料,合成了E)-3-(2-羟基-6-甲氧基苯基)-1-(10 -苯噻嗪-2-基)丙-烯-1-酮(3HM1P)。分析证实了3HM1P的分子结构,并得到了红外光谱的支持。采用B3LYP方法在6-311 ++G(d,p)水平对化合物的几何结构进行优化,并将理论计算与实验数据进行比较。通过线性光学吸收光谱分析,研究溶剂对3HM1P的影响,HOMO-LUMO分析显示,己烷中的能隙最高为3.27 eV。分子静电势(MEP)分析确定了富电子和缺电子区域,这是酶相互作用的关键。自然键轨道(NBO)分析表明,π→π *轨道重叠时的最大稳定能为392.59 kJ/mol。在早期药物相似性评估和ADMET虚拟筛选的支持下,通过对2JIU蛋白的分子对接研究来评估抗菌潜力。分子动力学模拟证实了3hmp1 - 3juv的成功对接,而正常模式分析表明,3hmp1 - 3juv在结合位点的分子迁移率稳定且灵活,特征值较低,为2.6383 × 10−04。本文章由计算机程序翻译,如有差异,请以英文原文为准。
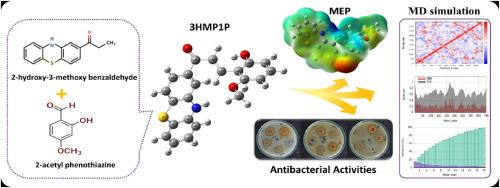
Synthesis, characterization and biological evaluation of a novel chalcone derivative: Spectroscopic, DFT, docking, ADMET and MD Studies
E)-3-(2-hydroxy-6-methoxyphenyl)-1-(1OH-Pheno thiazin-2-yl) prop 2-en-1-one (3HM1P) was synthesized by condensing 2-acetyl phenothiazine and 2-hydroxy-3-methoxy benzaldehyde. The molecular structure of 3HM1P was confirmed by analysis and well supported by FT-IR spectrum. The compound's geometry was optimized using the B3LYP method at the 6–311++G(d,p) level, and theoretical calculations were compared to experimental data. Linear optical absorption spectra were analyzed to study solvent effects on 3HM1P, with HOMO-LUMO analysis revealing a highest energy gap of 3.27 eV in hexane. Molecular electrostatic potential (MEP) analysis identified electron-rich and electron-deficient regions, crucial for enzyme interactions. Natural bond orbital (NBO) analysis indicated a maximum stabilization energy of 392.59 kJ/mol from π→π∗ orbital overlap. Antibacterial potential was assessed through molecular docking studies against the 2JIU protein, supported by early drug-likeness evaluations and ADMET virtual screening. Molecular dynamics simulations confirmed successful docking of 3HM1P-3JUV, while normal mode analysis indicated stable and flexible molecular mobility at the binding site, with a low eigenvalue of 2.6383 × 10−04.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


