速尿(形式I)在16种单溶剂中的研究:溶解度测定和模型、热力学分析、DFT计算和分子动力学模拟
IF 2.2
3区 工程技术
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
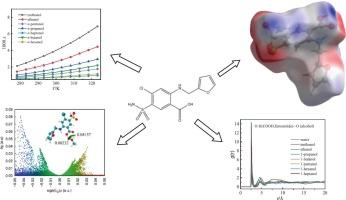
采用摇瓶饱和法,结合实验和计算方法,确定了速尿(I)在16种纯溶剂中的综合溶解度。随着温度的升高,速尿的溶解度增大。乙酸乙酯含量最高,水含量最低。x射线粉末衍射(XRD)扫描表明,在试验过程中没有观察到溶剂化和晶体转变。Apelblat方程提供了最好的相关结果。采用Wilson方程计算溶解热力学参数。此外,利用速尿胺分子表面的最小负静电电位和最大正静电电位来说明静电的酸碱特性。采用分子动力学模拟、密度泛函理论(DFT)计算和基于Hirshfeld分区的独立梯度分析,研究了速脲与溶剂的分子间相互作用。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Research on furosemide (form I) in sixteen mono-solvents: Solubility determination and models, thermodynamic analysis, DFT calculation and molecular dynamic simulation
The comprehensive solubilities of furosemide (I) in sixteen pure solvents were ascertained using the method of shake-flask saturation, together with experimental and computational approaches. The furosemide (I) solubility increased with rising temperature. It was highest in ethyl acetate, and lowest in water. No solvation, together with crystal transition, was observed during the trial process, as indicated by X-ray powder diffraction (XRD) scans. Apelblat equation provided the best correlation results. The Wilson equation was employed to calculate the dissolving thermodynamic parameters. Furthermore, the acidity-basicity characteristics of static electricity were illustrated utilizing the minimum negative and maximum positive electrostatic potential of the furosemide molecule surface. The intermolecular interactions of furosemide with solvents were examined using molecular dynamic simulation and density functional theory (DFT) calculations, and Hirshfeld partition-based independent gradient analysis.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Chemical Thermodynamics
工程技术-热力学
CiteScore
5.60
自引率
15.40%
发文量
199
审稿时长
79 days
期刊介绍:
The Journal of Chemical Thermodynamics exists primarily for dissemination of significant new knowledge in experimental equilibrium thermodynamics and transport properties of chemical systems. The defining attributes of The Journal are the quality and relevance of the papers published.
The Journal publishes work relating to gases, liquids, solids, polymers, mixtures, solutions and interfaces. Studies on systems with variability, such as biological or bio-based materials, gas hydrates, among others, will also be considered provided these are well characterized and reproducible where possible. Experimental methods should be described in sufficient detail to allow critical assessment of the accuracy claimed.
Authors are encouraged to provide physical or chemical interpretations of the results. Articles can contain modelling sections providing representations of data or molecular insights into the properties or transformations studied. Theoretical papers on chemical thermodynamics using molecular theory or modelling are also considered.
The Journal welcomes review articles in the field of chemical thermodynamics but prospective authors should first consult one of the Editors concerning the suitability of the proposed review.
Contributions of a routine nature or reporting on uncharacterised materials are not accepted.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


