{"title":"基于图的深度学习,整合单细胞和大量转录组数据,以识别临床癌症亚型。","authors":"Yixin Liu, Dandan Zhang, Tianyu Liu, Ao Wang, Guohua Wang, Yuming Zhao","doi":"10.1093/bib/bbaf467","DOIUrl":null,"url":null,"abstract":"<p><p>The integration of single-cell RNA sequencing (scRNA-seq) and bulk transcriptomic data has become essential for deciphering the complex heterogeneity of cancer and identifying clinical cancer subtypes. However, the inherent challenges posed by the high dimensionality, sparsity, and noise characteristics of scRNA-seq data have significantly hindered its widespread clinical translation. To address these limitations, we introduce single-cell and bulk transcriptomic graph deep learning, a graph-based deep learning method that synergistically integrates scRNA-seq and bulk transcriptomic data to precisely identify cancer subtypes and predict clinical outcomes. scBGDL constructs sample-specific gene graphs modeling complex gene-gene interactions and cellular relationships. The architecture employs Graph Attention Networks for feature aggregation, MinCutPool layers for dimensionality reduction, and Transformer modules to capture high-order biological dependencies. Independently validated in each of 16 distinct The Cancer Genome Atlas cancer types, scBGDL significantly outperformed existing methods in prognostic accuracy (mean C-index: 0.7060 versus 0.6709 max competitor), demonstrating robustness and generalizability to diverse transcriptional architectures. To demonstrate clinical versatility, we further evaluated scBGDL in three therapeutic contexts using multicenter cohorts: lung adenocarcinoma survival prediction (n = 1099), epithelial ovarian cancer platinum-based chemotherapy response (n = 762), skin cutaneous melanoma immunotherapy outcome (n = 305). scBGDL consistently delivered robust risk stratification (log-rank P < 0.05 across cohorts), identified key driver edges, and uncovered clinically relevant biological interpretations. By enabling multimodal data integration and interpretable biological insights, scBGDL advances precision oncology for prognosis prediction, therapy optimization, and biomarker discovery. The source code for scBGDL model is available online (https://github.com/NEFLab/scBGDL).</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"26 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12423395/pdf/","citationCount":"0","resultStr":"{\"title\":\"Graph-based deep learning for integrating single-cell and bulk transcriptomic data to identify clinical cancer subtypes.\",\"authors\":\"Yixin Liu, Dandan Zhang, Tianyu Liu, Ao Wang, Guohua Wang, Yuming Zhao\",\"doi\":\"10.1093/bib/bbaf467\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The integration of single-cell RNA sequencing (scRNA-seq) and bulk transcriptomic data has become essential for deciphering the complex heterogeneity of cancer and identifying clinical cancer subtypes. However, the inherent challenges posed by the high dimensionality, sparsity, and noise characteristics of scRNA-seq data have significantly hindered its widespread clinical translation. To address these limitations, we introduce single-cell and bulk transcriptomic graph deep learning, a graph-based deep learning method that synergistically integrates scRNA-seq and bulk transcriptomic data to precisely identify cancer subtypes and predict clinical outcomes. scBGDL constructs sample-specific gene graphs modeling complex gene-gene interactions and cellular relationships. The architecture employs Graph Attention Networks for feature aggregation, MinCutPool layers for dimensionality reduction, and Transformer modules to capture high-order biological dependencies. Independently validated in each of 16 distinct The Cancer Genome Atlas cancer types, scBGDL significantly outperformed existing methods in prognostic accuracy (mean C-index: 0.7060 versus 0.6709 max competitor), demonstrating robustness and generalizability to diverse transcriptional architectures. To demonstrate clinical versatility, we further evaluated scBGDL in three therapeutic contexts using multicenter cohorts: lung adenocarcinoma survival prediction (n = 1099), epithelial ovarian cancer platinum-based chemotherapy response (n = 762), skin cutaneous melanoma immunotherapy outcome (n = 305). scBGDL consistently delivered robust risk stratification (log-rank P < 0.05 across cohorts), identified key driver edges, and uncovered clinically relevant biological interpretations. By enabling multimodal data integration and interpretable biological insights, scBGDL advances precision oncology for prognosis prediction, therapy optimization, and biomarker discovery. The source code for scBGDL model is available online (https://github.com/NEFLab/scBGDL).</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"26 5\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2025-08-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12423395/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbaf467\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbaf467","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
摘要
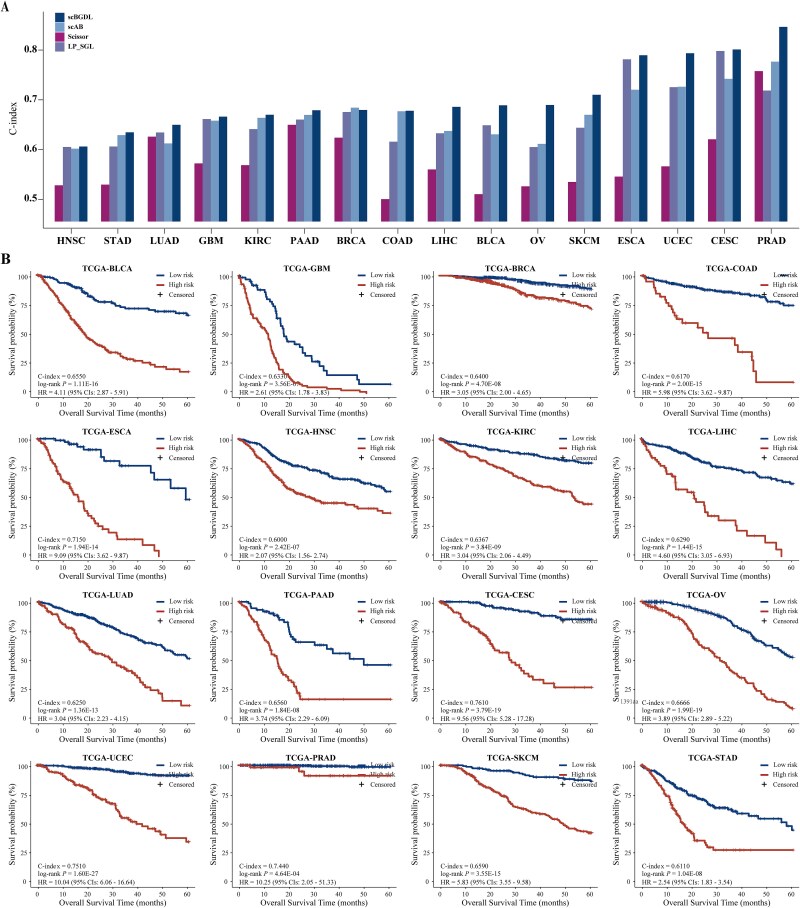
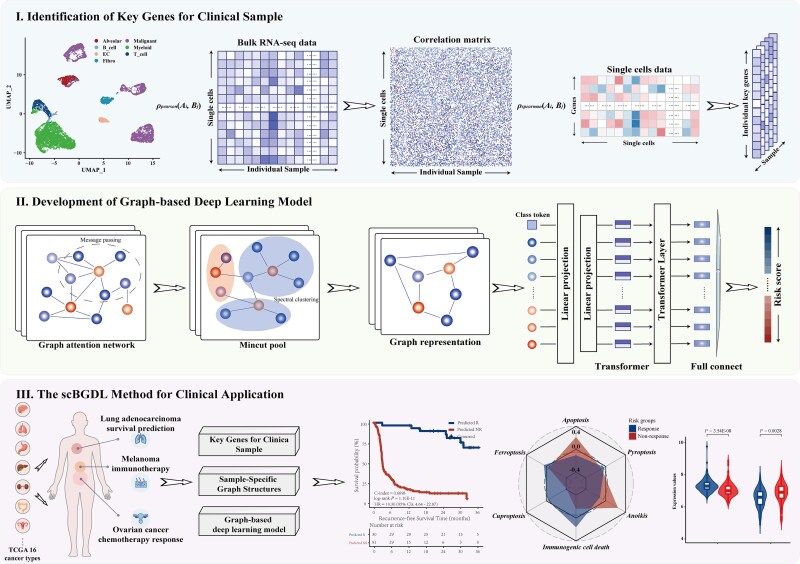
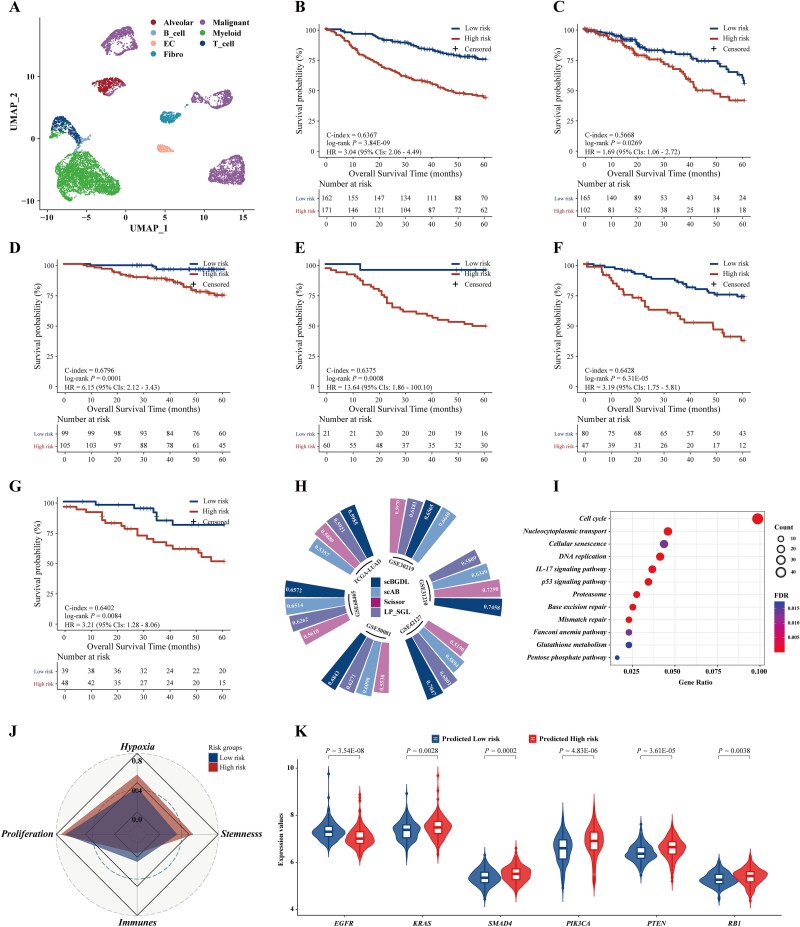
单细胞RNA测序(scRNA-seq)和大量转录组学数据的整合对于破译癌症的复杂异质性和识别临床癌症亚型至关重要。然而,scRNA-seq数据的高维性、稀疏性和噪声特征所带来的固有挑战极大地阻碍了其广泛的临床应用。为了解决这些限制,我们引入了单细胞和大量转录组图深度学习,这是一种基于图的深度学习方法,可以协同整合scRNA-seq和大量转录组数据,以精确识别癌症亚型并预测临床结果。scBGDL构建样本特异性基因图,模拟复杂的基因相互作用和细胞关系。该体系结构使用Graph Attention Networks进行特征聚合,使用MinCutPool层进行降维,使用Transformer模块捕获高阶生物依赖性。在16种不同的癌症基因组图谱癌症类型中独立验证,scBGDL在预后准确性方面明显优于现有方法(平均c指数:0.7060,最大竞争对手为0.6709),显示出鲁棒性和对不同转录结构的通用性。为了证明临床的多功能性,我们使用多中心队列进一步评估了三种治疗背景下的scBGDL:肺腺癌生存预测(n = 1099),上皮性卵巢癌铂基化疗反应(n = 762),皮肤皮肤黑色素瘤免疫治疗结果(n = 305)。scBGDL始终提供稳健的风险分层(log-rank P
Graph-based deep learning for integrating single-cell and bulk transcriptomic data to identify clinical cancer subtypes.
The integration of single-cell RNA sequencing (scRNA-seq) and bulk transcriptomic data has become essential for deciphering the complex heterogeneity of cancer and identifying clinical cancer subtypes. However, the inherent challenges posed by the high dimensionality, sparsity, and noise characteristics of scRNA-seq data have significantly hindered its widespread clinical translation. To address these limitations, we introduce single-cell and bulk transcriptomic graph deep learning, a graph-based deep learning method that synergistically integrates scRNA-seq and bulk transcriptomic data to precisely identify cancer subtypes and predict clinical outcomes. scBGDL constructs sample-specific gene graphs modeling complex gene-gene interactions and cellular relationships. The architecture employs Graph Attention Networks for feature aggregation, MinCutPool layers for dimensionality reduction, and Transformer modules to capture high-order biological dependencies. Independently validated in each of 16 distinct The Cancer Genome Atlas cancer types, scBGDL significantly outperformed existing methods in prognostic accuracy (mean C-index: 0.7060 versus 0.6709 max competitor), demonstrating robustness and generalizability to diverse transcriptional architectures. To demonstrate clinical versatility, we further evaluated scBGDL in three therapeutic contexts using multicenter cohorts: lung adenocarcinoma survival prediction (n = 1099), epithelial ovarian cancer platinum-based chemotherapy response (n = 762), skin cutaneous melanoma immunotherapy outcome (n = 305). scBGDL consistently delivered robust risk stratification (log-rank P < 0.05 across cohorts), identified key driver edges, and uncovered clinically relevant biological interpretations. By enabling multimodal data integration and interpretable biological insights, scBGDL advances precision oncology for prognosis prediction, therapy optimization, and biomarker discovery. The source code for scBGDL model is available online (https://github.com/NEFLab/scBGDL).
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


