Daniel Kaschta, Christina Post, Franziska Gaass, Milad Al-Tawil, Vincent Arriens, Saranya Balachandran, Tobias Bäumer, Valerie Berge, Friederike Birgel, Andreas Dalski, Maike Dittmar, Andre Franke, Sören Franzenburg, Janina Fuß, Bettina Gehring, Rebecca Gembicki, Bianca Greiten, Kristin Grohte, Britta Hanker, Kristian Händler, Lana Harder, Yorck Hellenbroich, Theresia Herget, Gloria Herrmann, Olaf Hiort, Kirstin Hoff, Birga Hoffmann, Nadine Hornig, Irina Hüning, Monika Kautza-Lucht, Juliane Köhler, Anna-Sophie Liegmann, Jasmin Lisfeld, Britt-Sabina Löscher, Nils G Margraf, Michelle Meyenborg, Anna Möllring, Hiltrud Muhle, Eva Maria Murga Penas, Henning Nommels, Dzhoy Papingi, Imke Poggenburg, Jelena Pozojevic, Philip Rosenstiel, Andreas Recke, Kimberly Roberts, Laelia Rösler, Franka Rust, Maj-Britt Salewski, Katharina Schau-Römer, Christian Schlein, Varun K A Sreenivasan, Louiza Toutouna, Caroline Utermann-Thüsing, Amelie T van der Ven, Alexander E Volk, Janne Wehnert, Sandra Wilson, Rixa Woitschach, Veronica Yumiceba, Christine Zühlke, Alexander Münchau, Norbert Brüggemann, Inga Vater, Almuth Caliebe, Inga Nagel, Malte Spielmann
{"title":"评估基因组测序策略:三人组、单例组和罕见病诊断中的标准检测。","authors":"Daniel Kaschta, Christina Post, Franziska Gaass, Milad Al-Tawil, Vincent Arriens, Saranya Balachandran, Tobias Bäumer, Valerie Berge, Friederike Birgel, Andreas Dalski, Maike Dittmar, Andre Franke, Sören Franzenburg, Janina Fuß, Bettina Gehring, Rebecca Gembicki, Bianca Greiten, Kristin Grohte, Britta Hanker, Kristian Händler, Lana Harder, Yorck Hellenbroich, Theresia Herget, Gloria Herrmann, Olaf Hiort, Kirstin Hoff, Birga Hoffmann, Nadine Hornig, Irina Hüning, Monika Kautza-Lucht, Juliane Köhler, Anna-Sophie Liegmann, Jasmin Lisfeld, Britt-Sabina Löscher, Nils G Margraf, Michelle Meyenborg, Anna Möllring, Hiltrud Muhle, Eva Maria Murga Penas, Henning Nommels, Dzhoy Papingi, Imke Poggenburg, Jelena Pozojevic, Philip Rosenstiel, Andreas Recke, Kimberly Roberts, Laelia Rösler, Franka Rust, Maj-Britt Salewski, Katharina Schau-Römer, Christian Schlein, Varun K A Sreenivasan, Louiza Toutouna, Caroline Utermann-Thüsing, Amelie T van der Ven, Alexander E Volk, Janne Wehnert, Sandra Wilson, Rixa Woitschach, Veronica Yumiceba, Christine Zühlke, Alexander Münchau, Norbert Brüggemann, Inga Vater, Almuth Caliebe, Inga Nagel, Malte Spielmann","doi":"10.1186/s13073-025-01516-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Short-read genome sequencing (GS) is among the most comprehensive genetic testing methods available, capable of detecting single-nucleotide variants, copy-number variants, mitochondrial variants, repeat expansions, and structural variants in a single assay. Despite its technical advantages, the full clinical utility of GS in real-world diagnostic settings remains to be fully established.</p><p><strong>Methods: </strong>This study systematically compared singleton GS (sGS), trio GS (tGS), and exome sequencing-based standard-of-care (SoC) genetic testing in 416 patients with rare diseases in a blinded, prospective study. Three independent teams with divergent baseline expertise evaluated the diagnostic yield of GS as a unifying first-tier test and directly compared its variant detection capabilities, learning curve, and clinical feasibility. The SoC team had extensive prior experience in exome-based diagnostics, while the sGS and tGS teams were newly trained in GS interpretation. Diagnostic yield was assessed through both prospective and retrospective analyses.</p><p><strong>Results: </strong>In our prospective analysis, tGS achieved the highest diagnostic yield for likely pathogenic/pathogenic variants at 36.1% in the newly trained team, surpassing the experienced SoC team at 35.1% and the newly trained sGS team at 28.8%. To evaluate which variants could technically be identified and account for differences in team experience, we conducted a retrospective analysis, achieving diagnostic yields of 36.7% for SoC, 39.1% for sGS, and 40.0% for tGS. The superior yield of GS was attributed to its ability to detect deep intronic, non-coding, and small copy-number variants missed by SoC. Notably, tGS identified three de novo variants classified as likely pathogenic based on recent GeneMatcher collaborations and newly published gene-disease association studies.</p><p><strong>Conclusions: </strong>Our findings demonstrate that GS, particularly tGS, outperforms SoC in diagnosing rare diseases, with sGS providing a more cost-effective alternative. These results suggest that GS should be considered a first-tier genetic test, offering an efficient, single-step approach to reduce the diagnostic odyssey for patients with rare diseases. The trio approach proved especially valuable for less experienced teams, as inheritance data facilitated variant interpretation and maintained high diagnostic yield, while experienced teams achieved comparable results with singleton analysis alone.</p>","PeriodicalId":12645,"journal":{"name":"Genome Medicine","volume":"17 1","pages":"100"},"PeriodicalIF":10.4000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12445032/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evaluating genome sequencing strategies: trio, singleton, and standard testing in rare disease diagnosis.\",\"authors\":\"Daniel Kaschta, Christina Post, Franziska Gaass, Milad Al-Tawil, Vincent Arriens, Saranya Balachandran, Tobias Bäumer, Valerie Berge, Friederike Birgel, Andreas Dalski, Maike Dittmar, Andre Franke, Sören Franzenburg, Janina Fuß, Bettina Gehring, Rebecca Gembicki, Bianca Greiten, Kristin Grohte, Britta Hanker, Kristian Händler, Lana Harder, Yorck Hellenbroich, Theresia Herget, Gloria Herrmann, Olaf Hiort, Kirstin Hoff, Birga Hoffmann, Nadine Hornig, Irina Hüning, Monika Kautza-Lucht, Juliane Köhler, Anna-Sophie Liegmann, Jasmin Lisfeld, Britt-Sabina Löscher, Nils G Margraf, Michelle Meyenborg, Anna Möllring, Hiltrud Muhle, Eva Maria Murga Penas, Henning Nommels, Dzhoy Papingi, Imke Poggenburg, Jelena Pozojevic, Philip Rosenstiel, Andreas Recke, Kimberly Roberts, Laelia Rösler, Franka Rust, Maj-Britt Salewski, Katharina Schau-Römer, Christian Schlein, Varun K A Sreenivasan, Louiza Toutouna, Caroline Utermann-Thüsing, Amelie T van der Ven, Alexander E Volk, Janne Wehnert, Sandra Wilson, Rixa Woitschach, Veronica Yumiceba, Christine Zühlke, Alexander Münchau, Norbert Brüggemann, Inga Vater, Almuth Caliebe, Inga Nagel, Malte Spielmann\",\"doi\":\"10.1186/s13073-025-01516-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Short-read genome sequencing (GS) is among the most comprehensive genetic testing methods available, capable of detecting single-nucleotide variants, copy-number variants, mitochondrial variants, repeat expansions, and structural variants in a single assay. Despite its technical advantages, the full clinical utility of GS in real-world diagnostic settings remains to be fully established.</p><p><strong>Methods: </strong>This study systematically compared singleton GS (sGS), trio GS (tGS), and exome sequencing-based standard-of-care (SoC) genetic testing in 416 patients with rare diseases in a blinded, prospective study. Three independent teams with divergent baseline expertise evaluated the diagnostic yield of GS as a unifying first-tier test and directly compared its variant detection capabilities, learning curve, and clinical feasibility. The SoC team had extensive prior experience in exome-based diagnostics, while the sGS and tGS teams were newly trained in GS interpretation. Diagnostic yield was assessed through both prospective and retrospective analyses.</p><p><strong>Results: </strong>In our prospective analysis, tGS achieved the highest diagnostic yield for likely pathogenic/pathogenic variants at 36.1% in the newly trained team, surpassing the experienced SoC team at 35.1% and the newly trained sGS team at 28.8%. To evaluate which variants could technically be identified and account for differences in team experience, we conducted a retrospective analysis, achieving diagnostic yields of 36.7% for SoC, 39.1% for sGS, and 40.0% for tGS. The superior yield of GS was attributed to its ability to detect deep intronic, non-coding, and small copy-number variants missed by SoC. Notably, tGS identified three de novo variants classified as likely pathogenic based on recent GeneMatcher collaborations and newly published gene-disease association studies.</p><p><strong>Conclusions: </strong>Our findings demonstrate that GS, particularly tGS, outperforms SoC in diagnosing rare diseases, with sGS providing a more cost-effective alternative. These results suggest that GS should be considered a first-tier genetic test, offering an efficient, single-step approach to reduce the diagnostic odyssey for patients with rare diseases. The trio approach proved especially valuable for less experienced teams, as inheritance data facilitated variant interpretation and maintained high diagnostic yield, while experienced teams achieved comparable results with singleton analysis alone.</p>\",\"PeriodicalId\":12645,\"journal\":{\"name\":\"Genome Medicine\",\"volume\":\"17 1\",\"pages\":\"100\"},\"PeriodicalIF\":10.4000,\"publicationDate\":\"2025-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12445032/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genome Medicine\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13073-025-01516-7\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genome Medicine","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13073-025-01516-7","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Evaluating genome sequencing strategies: trio, singleton, and standard testing in rare disease diagnosis.
Background: Short-read genome sequencing (GS) is among the most comprehensive genetic testing methods available, capable of detecting single-nucleotide variants, copy-number variants, mitochondrial variants, repeat expansions, and structural variants in a single assay. Despite its technical advantages, the full clinical utility of GS in real-world diagnostic settings remains to be fully established.
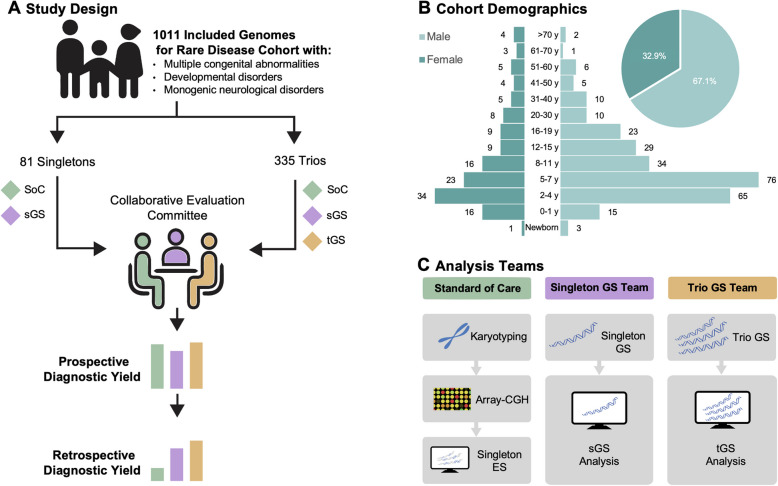
Methods: This study systematically compared singleton GS (sGS), trio GS (tGS), and exome sequencing-based standard-of-care (SoC) genetic testing in 416 patients with rare diseases in a blinded, prospective study. Three independent teams with divergent baseline expertise evaluated the diagnostic yield of GS as a unifying first-tier test and directly compared its variant detection capabilities, learning curve, and clinical feasibility. The SoC team had extensive prior experience in exome-based diagnostics, while the sGS and tGS teams were newly trained in GS interpretation. Diagnostic yield was assessed through both prospective and retrospective analyses.
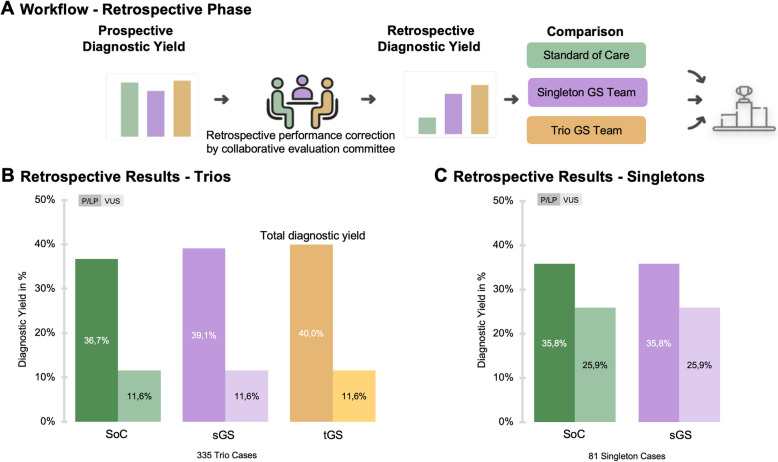
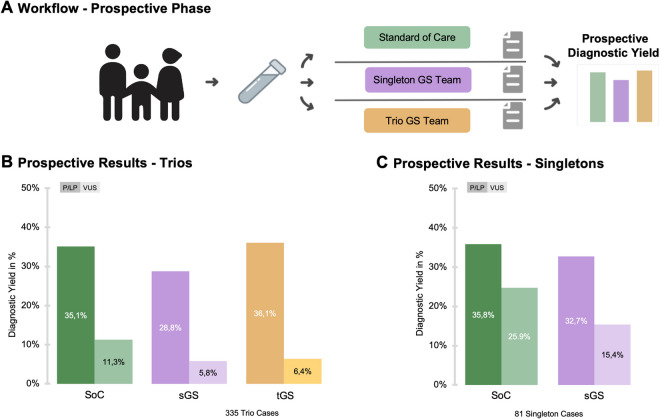
Results: In our prospective analysis, tGS achieved the highest diagnostic yield for likely pathogenic/pathogenic variants at 36.1% in the newly trained team, surpassing the experienced SoC team at 35.1% and the newly trained sGS team at 28.8%. To evaluate which variants could technically be identified and account for differences in team experience, we conducted a retrospective analysis, achieving diagnostic yields of 36.7% for SoC, 39.1% for sGS, and 40.0% for tGS. The superior yield of GS was attributed to its ability to detect deep intronic, non-coding, and small copy-number variants missed by SoC. Notably, tGS identified three de novo variants classified as likely pathogenic based on recent GeneMatcher collaborations and newly published gene-disease association studies.
Conclusions: Our findings demonstrate that GS, particularly tGS, outperforms SoC in diagnosing rare diseases, with sGS providing a more cost-effective alternative. These results suggest that GS should be considered a first-tier genetic test, offering an efficient, single-step approach to reduce the diagnostic odyssey for patients with rare diseases. The trio approach proved especially valuable for less experienced teams, as inheritance data facilitated variant interpretation and maintained high diagnostic yield, while experienced teams achieved comparable results with singleton analysis alone.
期刊介绍:
Genome Medicine is an open access journal that publishes outstanding research applying genetics, genomics, and multi-omics to understand, diagnose, and treat disease. Bridging basic science and clinical research, it covers areas such as cancer genomics, immuno-oncology, immunogenomics, infectious disease, microbiome, neurogenomics, systems medicine, clinical genomics, gene therapies, precision medicine, and clinical trials. The journal publishes original research, methods, software, and reviews to serve authors and promote broad interest and importance in the field.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


