Zafer Kandemir, , , Claudia Cardoso, , , Pino D’Amico, , , Cem Sevik*, , and , Kürşat Şendur*,
{"title":"从绝缘体到金属:用多体第一原理方法对二氧化钒光学性质的理论评价","authors":"Zafer Kandemir, , , Claudia Cardoso, , , Pino D’Amico, , , Cem Sevik*, , and , Kürşat Şendur*, ","doi":"10.1021/acs.jpcc.5c03003","DOIUrl":null,"url":null,"abstract":"<p >Vanadium dioxide (VO<sub>2</sub>) exhibits a temperature-driven insulator-to-metal transition, making it a promising material for optical and electronic applications. In this study, we perform a systematic first-principles investigation of the electronic and optical properties of VO<sub>2</sub> in its monoclinic (M<sub>1</sub>) and rutile (R) phases using density functional theory (DFT), many-body perturbation theory (G<sub>0</sub>W<sub>0</sub>), and the Bethe-Salpeter equation (BSE). Our results reveal that excitonic effects play a crucial role in accurately describing the dielectric response of the semiconducting M<sub>1</sub> phase, with G<sub>0</sub>W<sub>0</sub>/BSE and PBE/BSE approaches yielding optical spectra in excellent agreement with experimental data. For the metallic R phase, we find that the random phase approximation (RPA) at the PBE level provides a reliable description of its optical properties, particularly in the visible range, as long as intraband contributions are included. The frequency-dependent dielectric functions presented in this work achieve the required accuracy for large-scale optical simulations relevant to smart coatings and tunable infrared devices. To support further research and applications, we provide our computed optical data in an open-access repository on ZENODO.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 38","pages":"17202–17210"},"PeriodicalIF":3.2000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"From Insulator to Metal: Theoretical Assessment on the Optical Properties of Vanadium Dioxide Using Many-Body First-Principles Approaches\",\"authors\":\"Zafer Kandemir, , , Claudia Cardoso, , , Pino D’Amico, , , Cem Sevik*, , and , Kürşat Şendur*, \",\"doi\":\"10.1021/acs.jpcc.5c03003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Vanadium dioxide (VO<sub>2</sub>) exhibits a temperature-driven insulator-to-metal transition, making it a promising material for optical and electronic applications. In this study, we perform a systematic first-principles investigation of the electronic and optical properties of VO<sub>2</sub> in its monoclinic (M<sub>1</sub>) and rutile (R) phases using density functional theory (DFT), many-body perturbation theory (G<sub>0</sub>W<sub>0</sub>), and the Bethe-Salpeter equation (BSE). Our results reveal that excitonic effects play a crucial role in accurately describing the dielectric response of the semiconducting M<sub>1</sub> phase, with G<sub>0</sub>W<sub>0</sub>/BSE and PBE/BSE approaches yielding optical spectra in excellent agreement with experimental data. For the metallic R phase, we find that the random phase approximation (RPA) at the PBE level provides a reliable description of its optical properties, particularly in the visible range, as long as intraband contributions are included. The frequency-dependent dielectric functions presented in this work achieve the required accuracy for large-scale optical simulations relevant to smart coatings and tunable infrared devices. To support further research and applications, we provide our computed optical data in an open-access repository on ZENODO.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 38\",\"pages\":\"17202–17210\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03003\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03003","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
From Insulator to Metal: Theoretical Assessment on the Optical Properties of Vanadium Dioxide Using Many-Body First-Principles Approaches
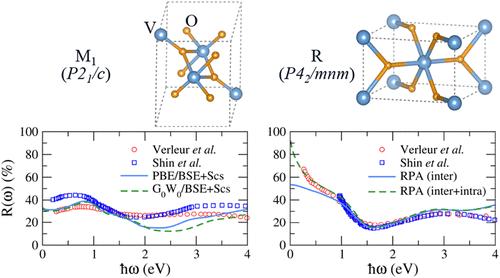
Vanadium dioxide (VO2) exhibits a temperature-driven insulator-to-metal transition, making it a promising material for optical and electronic applications. In this study, we perform a systematic first-principles investigation of the electronic and optical properties of VO2 in its monoclinic (M1) and rutile (R) phases using density functional theory (DFT), many-body perturbation theory (G0W0), and the Bethe-Salpeter equation (BSE). Our results reveal that excitonic effects play a crucial role in accurately describing the dielectric response of the semiconducting M1 phase, with G0W0/BSE and PBE/BSE approaches yielding optical spectra in excellent agreement with experimental data. For the metallic R phase, we find that the random phase approximation (RPA) at the PBE level provides a reliable description of its optical properties, particularly in the visible range, as long as intraband contributions are included. The frequency-dependent dielectric functions presented in this work achieve the required accuracy for large-scale optical simulations relevant to smart coatings and tunable infrared devices. To support further research and applications, we provide our computed optical data in an open-access repository on ZENODO.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


