Shahid Salam Bhat, , , Anjumun Rasool, , and , Manzoor Ahmad Dar*,
{"title":"有效二氧化碳转化的单原子功能化共价有机框架:来自第一原理模拟的电子结构见解","authors":"Shahid Salam Bhat, , , Anjumun Rasool, , and , Manzoor Ahmad Dar*, ","doi":"10.1021/acs.jpcc.5c04099","DOIUrl":null,"url":null,"abstract":"<p >Designing highly efficient catalysts with tunable reactivity and activity for the selective reduction of CO<sub>2</sub> is a challenging task that necessitates a thorough understanding of the catalyst electronic structure. In this work, we aim at systematically understanding the impact of single-atom functionalization on the electronic structure and CO<sub>2</sub> reduction activity of a hydrazine-based covalent organic framework (HCOF) using first-principles simulations. Our results demonstrate that the hydrazine linkages in the covalent organic framework (COF) are adequate for stabilizing a range of single atoms, resulting in a flexible electronic structure for successful activation of the centrosymmetric CO<sub>2</sub> molecule. We show that the single-atom-functionalized HCOF (SA-HCOF) systems bind the CO<sub>2</sub> molecule strongly in a selective manner with very high binding energies of −0.38 to −2.98 eV. In addition, through rigorous electronic structure analysis encompassing the distribution of d-states near the Fermi level and Bader charge analysis, we establish robust correlations between the CO<sub>2</sub> binding energy and the key electronic properties of the catalysts. The computed reaction pathways indicate that the Cr- and Co-based single-atom catalysts (SACs) show remarkable activity for CO<sub>2</sub> reduction to CO and HCOOH with very low limiting potentials of −0.61 and −0.52 V, respectively. Further, the CO<sub>2</sub> reduction activity of the COF-stabilized SACs was successfully correlated to the adsorption free energy of CO and HCOO intermediates which in turn depend on electronic properties such as the net Bader charge accumulated on the CO<sub>2</sub> molecule and the d-band center of the isolated metal atoms. These findings underscore the pivotal role of the electronic structure of isolated metal atoms stabilized on COFs in modulating the CO<sub>2</sub> reactivity and reduction activity, thereby providing crucial insights for the rational design of high-performance catalysts for CO<sub>2</sub> utilization.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 39","pages":"17512–17518"},"PeriodicalIF":3.2000,"publicationDate":"2025-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Single-Atom-Functionalized Covalent Organic Frameworks for Efficient CO2 Conversion: Electronic Structure Insights from First-Principles Simulations\",\"authors\":\"Shahid Salam Bhat, , , Anjumun Rasool, , and , Manzoor Ahmad Dar*, \",\"doi\":\"10.1021/acs.jpcc.5c04099\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Designing highly efficient catalysts with tunable reactivity and activity for the selective reduction of CO<sub>2</sub> is a challenging task that necessitates a thorough understanding of the catalyst electronic structure. In this work, we aim at systematically understanding the impact of single-atom functionalization on the electronic structure and CO<sub>2</sub> reduction activity of a hydrazine-based covalent organic framework (HCOF) using first-principles simulations. Our results demonstrate that the hydrazine linkages in the covalent organic framework (COF) are adequate for stabilizing a range of single atoms, resulting in a flexible electronic structure for successful activation of the centrosymmetric CO<sub>2</sub> molecule. We show that the single-atom-functionalized HCOF (SA-HCOF) systems bind the CO<sub>2</sub> molecule strongly in a selective manner with very high binding energies of −0.38 to −2.98 eV. In addition, through rigorous electronic structure analysis encompassing the distribution of d-states near the Fermi level and Bader charge analysis, we establish robust correlations between the CO<sub>2</sub> binding energy and the key electronic properties of the catalysts. The computed reaction pathways indicate that the Cr- and Co-based single-atom catalysts (SACs) show remarkable activity for CO<sub>2</sub> reduction to CO and HCOOH with very low limiting potentials of −0.61 and −0.52 V, respectively. Further, the CO<sub>2</sub> reduction activity of the COF-stabilized SACs was successfully correlated to the adsorption free energy of CO and HCOO intermediates which in turn depend on electronic properties such as the net Bader charge accumulated on the CO<sub>2</sub> molecule and the d-band center of the isolated metal atoms. These findings underscore the pivotal role of the electronic structure of isolated metal atoms stabilized on COFs in modulating the CO<sub>2</sub> reactivity and reduction activity, thereby providing crucial insights for the rational design of high-performance catalysts for CO<sub>2</sub> utilization.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 39\",\"pages\":\"17512–17518\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c04099\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c04099","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
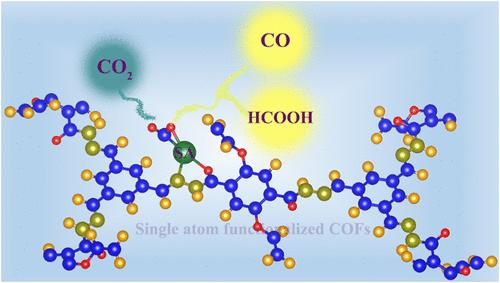
Single-Atom-Functionalized Covalent Organic Frameworks for Efficient CO2 Conversion: Electronic Structure Insights from First-Principles Simulations
Designing highly efficient catalysts with tunable reactivity and activity for the selective reduction of CO2 is a challenging task that necessitates a thorough understanding of the catalyst electronic structure. In this work, we aim at systematically understanding the impact of single-atom functionalization on the electronic structure and CO2 reduction activity of a hydrazine-based covalent organic framework (HCOF) using first-principles simulations. Our results demonstrate that the hydrazine linkages in the covalent organic framework (COF) are adequate for stabilizing a range of single atoms, resulting in a flexible electronic structure for successful activation of the centrosymmetric CO2 molecule. We show that the single-atom-functionalized HCOF (SA-HCOF) systems bind the CO2 molecule strongly in a selective manner with very high binding energies of −0.38 to −2.98 eV. In addition, through rigorous electronic structure analysis encompassing the distribution of d-states near the Fermi level and Bader charge analysis, we establish robust correlations between the CO2 binding energy and the key electronic properties of the catalysts. The computed reaction pathways indicate that the Cr- and Co-based single-atom catalysts (SACs) show remarkable activity for CO2 reduction to CO and HCOOH with very low limiting potentials of −0.61 and −0.52 V, respectively. Further, the CO2 reduction activity of the COF-stabilized SACs was successfully correlated to the adsorption free energy of CO and HCOO intermediates which in turn depend on electronic properties such as the net Bader charge accumulated on the CO2 molecule and the d-band center of the isolated metal atoms. These findings underscore the pivotal role of the electronic structure of isolated metal atoms stabilized on COFs in modulating the CO2 reactivity and reduction activity, thereby providing crucial insights for the rational design of high-performance catalysts for CO2 utilization.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


