{"title":"7-羟基喹啉基态和激发态质子转移反应中氢核量子效应的多组分密度泛函数研究","authors":"Taro Udagawa, Hinata Nagasaka, Yusuke Kanematsu, Takayoshi Ishimoto and Masanori Tachikawa","doi":"10.1039/D5CP02666K","DOIUrl":null,"url":null,"abstract":"<p >The proton transfer (PT) reaction in 7-hydroxyquinoline (7-HQ), mediated by three methanol molecules, has been investigated using time-dependent density functional theory (TD-DFT) and multi-component DFT (MC_TD-DFT) calculations, which can incorporate nuclear quantum effects (NQEs) of protons and deuterons. The NQEs were found to induce the geometrical changes in both the ground-state and excited-state, and alter orbital energies, affecting the HOMO–LUMO energy gap and absorption and fluorescence properties. The MC_DFT calculations predict a Stokes shift of 217 nm, closer to the experimental value (180–200 nm) compared to the conventional DFT (242 nm). For 7-HQ, the NQEs induced red shifts in absorption peaks and blue shifts in fluorescence peaks, aligning the Stokes shift more closely with experimental data. In addition, the MC_DFT calculations revealed that geometrical relaxation induced by the NQEs can be attributed to the shifts in the peaks in the case of 7-HQ. This study highlights the critical role of NQEs in understanding PT mechanisms, absorption and fluorescence properties, and H/D isotope effects, demonstrating the importance of including NQEs for accurate theoretical modeling.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 38","pages":" 20493-20499"},"PeriodicalIF":2.9000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A multi-component density functional study on quantum effects of hydrogen nuclei on ground-state and excited-state proton transfer reactions in 7-hydroxyquinoline\",\"authors\":\"Taro Udagawa, Hinata Nagasaka, Yusuke Kanematsu, Takayoshi Ishimoto and Masanori Tachikawa\",\"doi\":\"10.1039/D5CP02666K\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The proton transfer (PT) reaction in 7-hydroxyquinoline (7-HQ), mediated by three methanol molecules, has been investigated using time-dependent density functional theory (TD-DFT) and multi-component DFT (MC_TD-DFT) calculations, which can incorporate nuclear quantum effects (NQEs) of protons and deuterons. The NQEs were found to induce the geometrical changes in both the ground-state and excited-state, and alter orbital energies, affecting the HOMO–LUMO energy gap and absorption and fluorescence properties. The MC_DFT calculations predict a Stokes shift of 217 nm, closer to the experimental value (180–200 nm) compared to the conventional DFT (242 nm). For 7-HQ, the NQEs induced red shifts in absorption peaks and blue shifts in fluorescence peaks, aligning the Stokes shift more closely with experimental data. In addition, the MC_DFT calculations revealed that geometrical relaxation induced by the NQEs can be attributed to the shifts in the peaks in the case of 7-HQ. This study highlights the critical role of NQEs in understanding PT mechanisms, absorption and fluorescence properties, and H/D isotope effects, demonstrating the importance of including NQEs for accurate theoretical modeling.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 38\",\"pages\":\" 20493-20499\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02666k\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02666k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A multi-component density functional study on quantum effects of hydrogen nuclei on ground-state and excited-state proton transfer reactions in 7-hydroxyquinoline
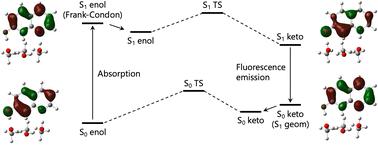
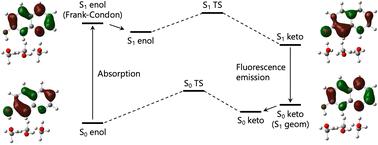
The proton transfer (PT) reaction in 7-hydroxyquinoline (7-HQ), mediated by three methanol molecules, has been investigated using time-dependent density functional theory (TD-DFT) and multi-component DFT (MC_TD-DFT) calculations, which can incorporate nuclear quantum effects (NQEs) of protons and deuterons. The NQEs were found to induce the geometrical changes in both the ground-state and excited-state, and alter orbital energies, affecting the HOMO–LUMO energy gap and absorption and fluorescence properties. The MC_DFT calculations predict a Stokes shift of 217 nm, closer to the experimental value (180–200 nm) compared to the conventional DFT (242 nm). For 7-HQ, the NQEs induced red shifts in absorption peaks and blue shifts in fluorescence peaks, aligning the Stokes shift more closely with experimental data. In addition, the MC_DFT calculations revealed that geometrical relaxation induced by the NQEs can be attributed to the shifts in the peaks in the case of 7-HQ. This study highlights the critical role of NQEs in understanding PT mechanisms, absorption and fluorescence properties, and H/D isotope effects, demonstrating the importance of including NQEs for accurate theoretical modeling.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


