{"title":"PH3的势能面及非谐波谱研究","authors":"Yunguang Zhang, Yingying Fang, Jingzao Cao, Youhan Cheng, Wenyu Xi, Qi Dong","doi":"10.1134/S0036024425701572","DOIUrl":null,"url":null,"abstract":"<p>The potential energy surface and vibration spectrum of the interstellar molecule Phosphine (PH<sub>3</sub>) are calculated ab initio by using the MOLPRO 2018 quantization program package based on the high-precision coupled CCSD(T)-F12a theoretical method and the cc-pVTZ-F12 basis set. Firstly, the equilibrium geometric structure of the PH<sub>3</sub> molecule is calculated by using the coupled clusters CCSD, CCSD(T), CCSD(T)-F12a, and the Møller–Plesset perturbation theory to second order (MP2) methods in combination with different basis sets, and it is found that the optimized structure obtained at the CCSD(T)-F12a/cc-pVTZ-F12 level is in agreement with the experimental values. In addition, there are potential wells that appear in the two-dimensional relative potential energy surface of the vibrational mode containing <i>q</i><sub>2</sub>, which is very different from the previously studied potential energy surface of the Phosphaethyne (HCP) molecule. Secondly, the vibrational multi-configuration self-consistent field (VMCSCF) and the vibrational multi-reference configuration interaction (VMRCI) are used to calculate the PH<sub>3</sub> fundamental frequencies, overtones, and combination frequencies, and found that the degenerate phenomenon between the fundamental frequencies and the Fermi resonance between overtones and combination frequencies. Finally, the infrared and Raman spectra are plotted using the calculated anharmonic frequencies, and the above phenomena can also be observed from the spectrogram. This paper provides a valuable reference for the study of interstellar molecules containing P.</p>","PeriodicalId":767,"journal":{"name":"Russian Journal of Physical Chemistry A","volume":"99 9","pages":"2127 - 2136"},"PeriodicalIF":0.8000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Research on the Potential Energy Surface and Anharmonic Spectra of the PH3\",\"authors\":\"Yunguang Zhang, Yingying Fang, Jingzao Cao, Youhan Cheng, Wenyu Xi, Qi Dong\",\"doi\":\"10.1134/S0036024425701572\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The potential energy surface and vibration spectrum of the interstellar molecule Phosphine (PH<sub>3</sub>) are calculated ab initio by using the MOLPRO 2018 quantization program package based on the high-precision coupled CCSD(T)-F12a theoretical method and the cc-pVTZ-F12 basis set. Firstly, the equilibrium geometric structure of the PH<sub>3</sub> molecule is calculated by using the coupled clusters CCSD, CCSD(T), CCSD(T)-F12a, and the Møller–Plesset perturbation theory to second order (MP2) methods in combination with different basis sets, and it is found that the optimized structure obtained at the CCSD(T)-F12a/cc-pVTZ-F12 level is in agreement with the experimental values. In addition, there are potential wells that appear in the two-dimensional relative potential energy surface of the vibrational mode containing <i>q</i><sub>2</sub>, which is very different from the previously studied potential energy surface of the Phosphaethyne (HCP) molecule. Secondly, the vibrational multi-configuration self-consistent field (VMCSCF) and the vibrational multi-reference configuration interaction (VMRCI) are used to calculate the PH<sub>3</sub> fundamental frequencies, overtones, and combination frequencies, and found that the degenerate phenomenon between the fundamental frequencies and the Fermi resonance between overtones and combination frequencies. Finally, the infrared and Raman spectra are plotted using the calculated anharmonic frequencies, and the above phenomena can also be observed from the spectrogram. This paper provides a valuable reference for the study of interstellar molecules containing P.</p>\",\"PeriodicalId\":767,\"journal\":{\"name\":\"Russian Journal of Physical Chemistry A\",\"volume\":\"99 9\",\"pages\":\"2127 - 2136\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2025-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Russian Journal of Physical Chemistry A\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S0036024425701572\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Russian Journal of Physical Chemistry A","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1134/S0036024425701572","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Research on the Potential Energy Surface and Anharmonic Spectra of the PH3
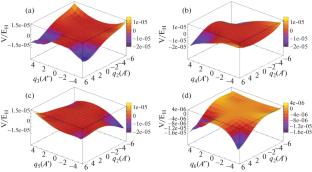
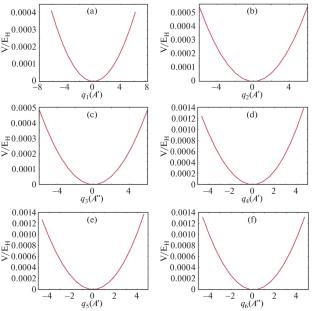
The potential energy surface and vibration spectrum of the interstellar molecule Phosphine (PH3) are calculated ab initio by using the MOLPRO 2018 quantization program package based on the high-precision coupled CCSD(T)-F12a theoretical method and the cc-pVTZ-F12 basis set. Firstly, the equilibrium geometric structure of the PH3 molecule is calculated by using the coupled clusters CCSD, CCSD(T), CCSD(T)-F12a, and the Møller–Plesset perturbation theory to second order (MP2) methods in combination with different basis sets, and it is found that the optimized structure obtained at the CCSD(T)-F12a/cc-pVTZ-F12 level is in agreement with the experimental values. In addition, there are potential wells that appear in the two-dimensional relative potential energy surface of the vibrational mode containing q2, which is very different from the previously studied potential energy surface of the Phosphaethyne (HCP) molecule. Secondly, the vibrational multi-configuration self-consistent field (VMCSCF) and the vibrational multi-reference configuration interaction (VMRCI) are used to calculate the PH3 fundamental frequencies, overtones, and combination frequencies, and found that the degenerate phenomenon between the fundamental frequencies and the Fermi resonance between overtones and combination frequencies. Finally, the infrared and Raman spectra are plotted using the calculated anharmonic frequencies, and the above phenomena can also be observed from the spectrogram. This paper provides a valuable reference for the study of interstellar molecules containing P.
期刊介绍:
Russian Journal of Physical Chemistry A. Focus on Chemistry (Zhurnal Fizicheskoi Khimii), founded in 1930, offers a comprehensive review of theoretical and experimental research from the Russian Academy of Sciences, leading research and academic centers from Russia and from all over the world.
Articles are devoted to chemical thermodynamics and thermochemistry, biophysical chemistry, photochemistry and magnetochemistry, materials structure, quantum chemistry, physical chemistry of nanomaterials and solutions, surface phenomena and adsorption, and methods and techniques of physicochemical studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


