Ji Peng, Ting Huang, Qiulan Wang, Bijing Liu, Luzhen Qin, Yuanyuan Yao
{"title":"努南综合征伴PTPN11基因变异,表现为孤立性身材矮小1例。","authors":"Ji Peng, Ting Huang, Qiulan Wang, Bijing Liu, Luzhen Qin, Yuanyuan Yao","doi":"10.21037/tp-2025-422","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Noonan syndrome (NS) is an autosomal dominant genetic disorder characterized by significant clinical heterogeneity, including distinctive facial features, short stature, developmental delay, congenital heart defects, and thoracic deformities. In some patients, especially in the early stages, the clinical manifestations are atypical and may solely include growth retardation or short stature, leading to misdiagnosis as idiopathic short stature or growth hormone (GH) deficiency and thereby delaying timely intervention.</p><p><strong>Case description: </strong>In this report, a female child is described who exhibited slow weight and height gain since birth. At 18 months, physical examination revealed that her height was below the 3rd percentile, with bone age delayed by 3 months. No typical NS facial features or congenital heart defects were observed. GH stimulation testing indicated partial GH deficiency, and the insulin-like growth factor 1 (IGF-1) level was below normal; thus, a preliminary diagnosis of short stature due to GH insufficiency was made, and nutritional intervention was recommended. However, follow-up revealed a persistent slow growth velocity and progressive bone age delay. At 6 years and 1 month of age, whole-exome sequencing identified a heterozygous c.923A>G (p.Asn308Ser) variant in the <i>PTPN11</i> gene, confirming the diagnosis of NS. Recombinant human growth hormone (rhGH) therapy was subsequently initiated. After rhGH intervention, the patient's annual height velocity increased by 2.4 to 3.2 times compared to pretreatment, which was accompanied by synchronous progression of bone age.</p><p><strong>Conclusions: </strong>This case highlights the importance of early genetic testing in patients with persistent growth retardation unresponsive to standard interventions. rhGH therapy has been shown to be safe and effective in patients with NS and short stature, significantly promoting height gain and bone age development, and should be considered for broader clinical application.</p>","PeriodicalId":23294,"journal":{"name":"Translational pediatrics","volume":"14 8","pages":"2057-2065"},"PeriodicalIF":1.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12433046/pdf/","citationCount":"0","resultStr":"{\"title\":\"Noonan syndrome with <i>PTPN11</i> gene variant presenting as isolated short stature: a case report.\",\"authors\":\"Ji Peng, Ting Huang, Qiulan Wang, Bijing Liu, Luzhen Qin, Yuanyuan Yao\",\"doi\":\"10.21037/tp-2025-422\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Noonan syndrome (NS) is an autosomal dominant genetic disorder characterized by significant clinical heterogeneity, including distinctive facial features, short stature, developmental delay, congenital heart defects, and thoracic deformities. In some patients, especially in the early stages, the clinical manifestations are atypical and may solely include growth retardation or short stature, leading to misdiagnosis as idiopathic short stature or growth hormone (GH) deficiency and thereby delaying timely intervention.</p><p><strong>Case description: </strong>In this report, a female child is described who exhibited slow weight and height gain since birth. At 18 months, physical examination revealed that her height was below the 3rd percentile, with bone age delayed by 3 months. No typical NS facial features or congenital heart defects were observed. GH stimulation testing indicated partial GH deficiency, and the insulin-like growth factor 1 (IGF-1) level was below normal; thus, a preliminary diagnosis of short stature due to GH insufficiency was made, and nutritional intervention was recommended. However, follow-up revealed a persistent slow growth velocity and progressive bone age delay. At 6 years and 1 month of age, whole-exome sequencing identified a heterozygous c.923A>G (p.Asn308Ser) variant in the <i>PTPN11</i> gene, confirming the diagnosis of NS. Recombinant human growth hormone (rhGH) therapy was subsequently initiated. After rhGH intervention, the patient's annual height velocity increased by 2.4 to 3.2 times compared to pretreatment, which was accompanied by synchronous progression of bone age.</p><p><strong>Conclusions: </strong>This case highlights the importance of early genetic testing in patients with persistent growth retardation unresponsive to standard interventions. rhGH therapy has been shown to be safe and effective in patients with NS and short stature, significantly promoting height gain and bone age development, and should be considered for broader clinical application.</p>\",\"PeriodicalId\":23294,\"journal\":{\"name\":\"Translational pediatrics\",\"volume\":\"14 8\",\"pages\":\"2057-2065\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-08-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12433046/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational pediatrics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tp-2025-422\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tp-2025-422","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
Noonan syndrome with PTPN11 gene variant presenting as isolated short stature: a case report.
Background: Noonan syndrome (NS) is an autosomal dominant genetic disorder characterized by significant clinical heterogeneity, including distinctive facial features, short stature, developmental delay, congenital heart defects, and thoracic deformities. In some patients, especially in the early stages, the clinical manifestations are atypical and may solely include growth retardation or short stature, leading to misdiagnosis as idiopathic short stature or growth hormone (GH) deficiency and thereby delaying timely intervention.
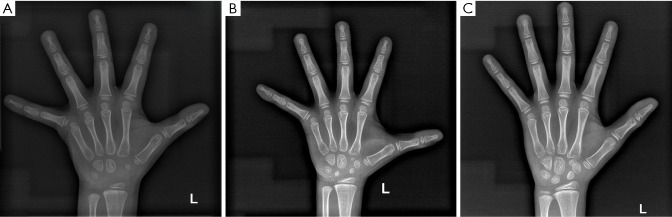
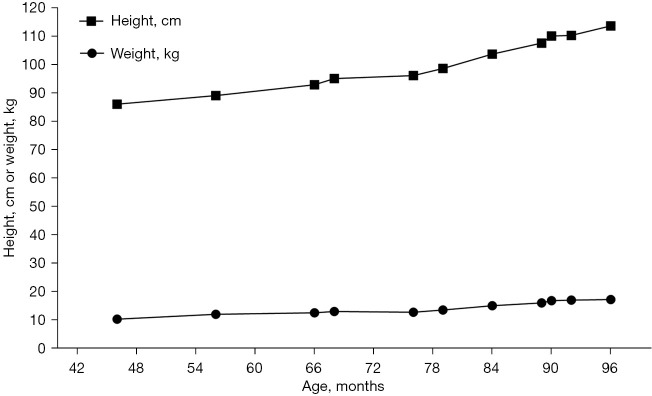
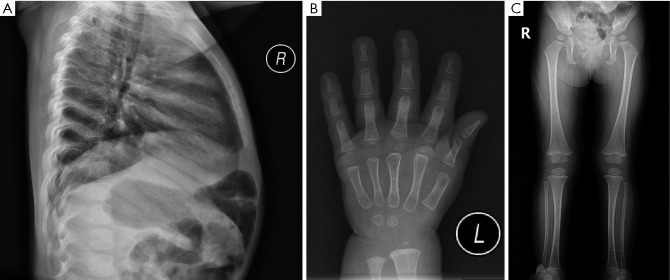
Case description: In this report, a female child is described who exhibited slow weight and height gain since birth. At 18 months, physical examination revealed that her height was below the 3rd percentile, with bone age delayed by 3 months. No typical NS facial features or congenital heart defects were observed. GH stimulation testing indicated partial GH deficiency, and the insulin-like growth factor 1 (IGF-1) level was below normal; thus, a preliminary diagnosis of short stature due to GH insufficiency was made, and nutritional intervention was recommended. However, follow-up revealed a persistent slow growth velocity and progressive bone age delay. At 6 years and 1 month of age, whole-exome sequencing identified a heterozygous c.923A>G (p.Asn308Ser) variant in the PTPN11 gene, confirming the diagnosis of NS. Recombinant human growth hormone (rhGH) therapy was subsequently initiated. After rhGH intervention, the patient's annual height velocity increased by 2.4 to 3.2 times compared to pretreatment, which was accompanied by synchronous progression of bone age.
Conclusions: This case highlights the importance of early genetic testing in patients with persistent growth retardation unresponsive to standard interventions. rhGH therapy has been shown to be safe and effective in patients with NS and short stature, significantly promoting height gain and bone age development, and should be considered for broader clinical application.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


