Wan Huai, Juan Li, Xin Li, Yu Ding, Tingting Yu, Hao Zhang, Xiumin Wang, Ruen Yao
{"title":"Weiss-Kruszka综合征患者ZNF462的新变异和表型更新:一个病例系列。","authors":"Wan Huai, Juan Li, Xin Li, Yu Ding, Tingting Yu, Hao Zhang, Xiumin Wang, Ruen Yao","doi":"10.21037/tp-2025-274","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Weiss-Kruszka syndrome (WSKA) is a neurodevelopmental disorder caused by loss-of-function variants in <i>ZNF462</i>, a zinc-finger transcription factor critical for embryonic morphogenesis. Characterized by intellectual disability, facial dysmorphism, muscle tone abnormalities, and congenital malformations, WSKA exhibits significant phenotypic heterogeneity. Despite over 40 cases reported globally, existing studies rarely report the clinical manifestations of this disease in Chinese patients.</p><p><strong>Case description: </strong>Six patients harboring pathogenic <i>ZNF462</i> variants were included and clinically evaluated at Shanghai Children's Medical Center. Comprehensive phenotyping included detailed dysmorphology assessments, neurodevelopmental testing, and targeted organ system evaluations. Whole-exome sequencing (WES) with Sanger confirmation identified pathogenic <i>ZNF462</i> variants. Additionally, phenotypic data from prior published cases were systematically reviewed. WES confirmed the diagnosis of WSKA in six patients from five pedigrees, revealing five novel <i>ZNF462</i> variants. Phenotypic information in our Chinese patient cohort revealed similar neurodevelopmental abnormalities and dysmorphic facial features. Other clinical features, including growth hormone deficiency, limb anomalies, and abnormal gonadal development, were also observed within different individuals in our cohort, suggesting phenotypic variability and heterogeneity among different ethnic origins.</p><p><strong>Conclusions: </strong>Identifying novel pathogenic variants in <i>ZNF462</i> and compiling a comprehensive clinical phenotype spectrum of patients could provide crucial information for the precise diagnosis and treatment of WSKA in Han Chinese individuals. Our findings underscore the necessity of ethnicity-specific diagnostic criteria.</p>","PeriodicalId":23294,"journal":{"name":"Translational pediatrics","volume":"14 8","pages":"1991-2000"},"PeriodicalIF":1.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12433108/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel variants in <i>ZNF462</i> and phenotype update in patients with Weiss-Kruszka syndrome: a case series.\",\"authors\":\"Wan Huai, Juan Li, Xin Li, Yu Ding, Tingting Yu, Hao Zhang, Xiumin Wang, Ruen Yao\",\"doi\":\"10.21037/tp-2025-274\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Weiss-Kruszka syndrome (WSKA) is a neurodevelopmental disorder caused by loss-of-function variants in <i>ZNF462</i>, a zinc-finger transcription factor critical for embryonic morphogenesis. Characterized by intellectual disability, facial dysmorphism, muscle tone abnormalities, and congenital malformations, WSKA exhibits significant phenotypic heterogeneity. Despite over 40 cases reported globally, existing studies rarely report the clinical manifestations of this disease in Chinese patients.</p><p><strong>Case description: </strong>Six patients harboring pathogenic <i>ZNF462</i> variants were included and clinically evaluated at Shanghai Children's Medical Center. Comprehensive phenotyping included detailed dysmorphology assessments, neurodevelopmental testing, and targeted organ system evaluations. Whole-exome sequencing (WES) with Sanger confirmation identified pathogenic <i>ZNF462</i> variants. Additionally, phenotypic data from prior published cases were systematically reviewed. WES confirmed the diagnosis of WSKA in six patients from five pedigrees, revealing five novel <i>ZNF462</i> variants. Phenotypic information in our Chinese patient cohort revealed similar neurodevelopmental abnormalities and dysmorphic facial features. Other clinical features, including growth hormone deficiency, limb anomalies, and abnormal gonadal development, were also observed within different individuals in our cohort, suggesting phenotypic variability and heterogeneity among different ethnic origins.</p><p><strong>Conclusions: </strong>Identifying novel pathogenic variants in <i>ZNF462</i> and compiling a comprehensive clinical phenotype spectrum of patients could provide crucial information for the precise diagnosis and treatment of WSKA in Han Chinese individuals. Our findings underscore the necessity of ethnicity-specific diagnostic criteria.</p>\",\"PeriodicalId\":23294,\"journal\":{\"name\":\"Translational pediatrics\",\"volume\":\"14 8\",\"pages\":\"1991-2000\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-08-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12433108/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational pediatrics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tp-2025-274\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tp-2025-274","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/20 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
Novel variants in ZNF462 and phenotype update in patients with Weiss-Kruszka syndrome: a case series.
Background: Weiss-Kruszka syndrome (WSKA) is a neurodevelopmental disorder caused by loss-of-function variants in ZNF462, a zinc-finger transcription factor critical for embryonic morphogenesis. Characterized by intellectual disability, facial dysmorphism, muscle tone abnormalities, and congenital malformations, WSKA exhibits significant phenotypic heterogeneity. Despite over 40 cases reported globally, existing studies rarely report the clinical manifestations of this disease in Chinese patients.
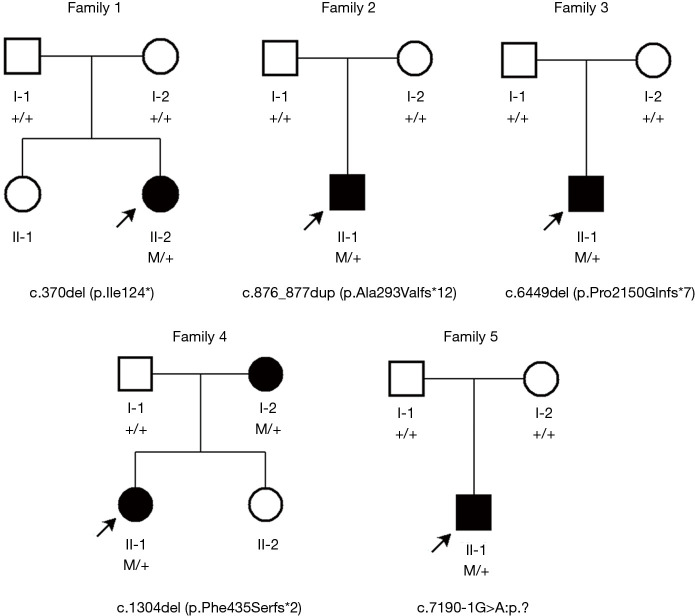
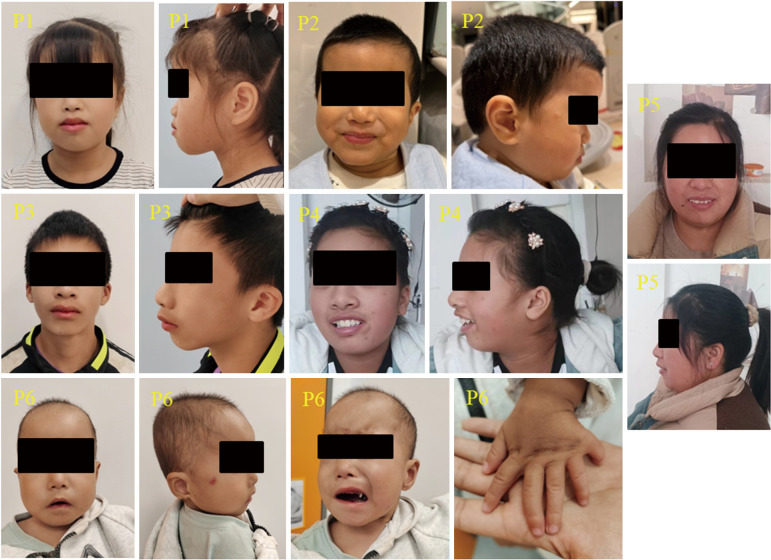
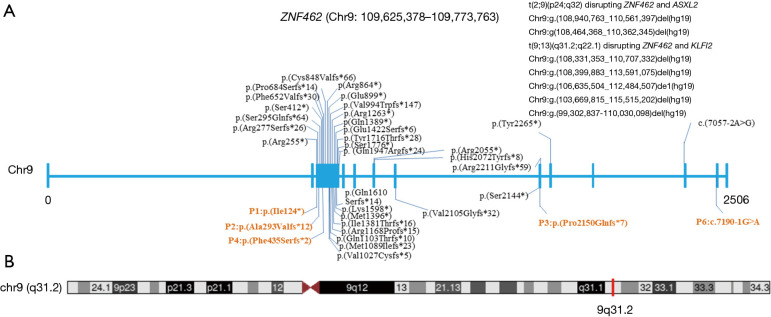
Case description: Six patients harboring pathogenic ZNF462 variants were included and clinically evaluated at Shanghai Children's Medical Center. Comprehensive phenotyping included detailed dysmorphology assessments, neurodevelopmental testing, and targeted organ system evaluations. Whole-exome sequencing (WES) with Sanger confirmation identified pathogenic ZNF462 variants. Additionally, phenotypic data from prior published cases were systematically reviewed. WES confirmed the diagnosis of WSKA in six patients from five pedigrees, revealing five novel ZNF462 variants. Phenotypic information in our Chinese patient cohort revealed similar neurodevelopmental abnormalities and dysmorphic facial features. Other clinical features, including growth hormone deficiency, limb anomalies, and abnormal gonadal development, were also observed within different individuals in our cohort, suggesting phenotypic variability and heterogeneity among different ethnic origins.
Conclusions: Identifying novel pathogenic variants in ZNF462 and compiling a comprehensive clinical phenotype spectrum of patients could provide crucial information for the precise diagnosis and treatment of WSKA in Han Chinese individuals. Our findings underscore the necessity of ethnicity-specific diagnostic criteria.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


