Anuj Kumar Ray, Sandeep Nagar, Girish Varma, U. Deva Priyakumar and Ankan Paul
{"title":"利用高自旋DFT特征预测三维过渡金属配合物中的自旋态间隙","authors":"Anuj Kumar Ray, Sandeep Nagar, Girish Varma, U. Deva Priyakumar and Ankan Paul","doi":"10.1039/D5CP02155C","DOIUrl":null,"url":null,"abstract":"<p >Determining spin state energy gaps (SSE) of 3d transition metal complexes (TMCs) is a major challenge in theoretical chemistry, as high-level quantum methods, though reliable, are computationally impractical for large-scale studies. This work explores a machine learning (ML)-based approach to predict DFT adiabatic SSE gaps using descriptors derived from a single high-spin DFT calculation. This approach is adopted to eliminate the differential treatment of electronic correlation between high-spin and low-spin structures. Our descriptors aim to incorporate the knowledge of crystal field theory into the ML model. They include atomic energy levels of bare metal ions, natural charges of ligating atoms, d-orbital molecular orbital eigenvalues derived from an high spin calculation, HOMO–LUMO gaps of free ligands, and simple identity-based features. We train ML models on 1434 SSE values spanning 934 complexes and demonstrate their transferability to more challenging complexes having bidentate π-bonding ligands despite being trained on simpler Werner-type monodentate complexes. We achieved a minimum MAE of 4.0 kcal mol<small><sup>−1</sup></small> on the monodentate test set, and maintained a comparable MAE of 6.6 kcal mol<small><sup>−1</sup></small> in the transferability assessment. This approach bypasses the need for multi-reference low-spin optimizations while retaining predictive accuracy, offering a cost-effective strategy for SSE estimation in transition metal chemistry. We hope the insights covered in this study will contribute to the development of additional electronic structure-based descriptors for SSE predictions.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 38","pages":" 20717-20725"},"PeriodicalIF":2.9000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Leveraging high-spin DFT features for prediction of spin state gaps in 3d transition metal complexes\",\"authors\":\"Anuj Kumar Ray, Sandeep Nagar, Girish Varma, U. Deva Priyakumar and Ankan Paul\",\"doi\":\"10.1039/D5CP02155C\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Determining spin state energy gaps (SSE) of 3d transition metal complexes (TMCs) is a major challenge in theoretical chemistry, as high-level quantum methods, though reliable, are computationally impractical for large-scale studies. This work explores a machine learning (ML)-based approach to predict DFT adiabatic SSE gaps using descriptors derived from a single high-spin DFT calculation. This approach is adopted to eliminate the differential treatment of electronic correlation between high-spin and low-spin structures. Our descriptors aim to incorporate the knowledge of crystal field theory into the ML model. They include atomic energy levels of bare metal ions, natural charges of ligating atoms, d-orbital molecular orbital eigenvalues derived from an high spin calculation, HOMO–LUMO gaps of free ligands, and simple identity-based features. We train ML models on 1434 SSE values spanning 934 complexes and demonstrate their transferability to more challenging complexes having bidentate π-bonding ligands despite being trained on simpler Werner-type monodentate complexes. We achieved a minimum MAE of 4.0 kcal mol<small><sup>−1</sup></small> on the monodentate test set, and maintained a comparable MAE of 6.6 kcal mol<small><sup>−1</sup></small> in the transferability assessment. This approach bypasses the need for multi-reference low-spin optimizations while retaining predictive accuracy, offering a cost-effective strategy for SSE estimation in transition metal chemistry. We hope the insights covered in this study will contribute to the development of additional electronic structure-based descriptors for SSE predictions.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 38\",\"pages\":\" 20717-20725\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02155c\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp02155c","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Leveraging high-spin DFT features for prediction of spin state gaps in 3d transition metal complexes
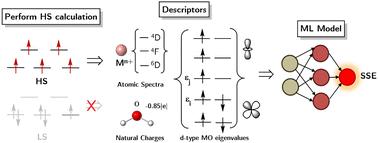
Determining spin state energy gaps (SSE) of 3d transition metal complexes (TMCs) is a major challenge in theoretical chemistry, as high-level quantum methods, though reliable, are computationally impractical for large-scale studies. This work explores a machine learning (ML)-based approach to predict DFT adiabatic SSE gaps using descriptors derived from a single high-spin DFT calculation. This approach is adopted to eliminate the differential treatment of electronic correlation between high-spin and low-spin structures. Our descriptors aim to incorporate the knowledge of crystal field theory into the ML model. They include atomic energy levels of bare metal ions, natural charges of ligating atoms, d-orbital molecular orbital eigenvalues derived from an high spin calculation, HOMO–LUMO gaps of free ligands, and simple identity-based features. We train ML models on 1434 SSE values spanning 934 complexes and demonstrate their transferability to more challenging complexes having bidentate π-bonding ligands despite being trained on simpler Werner-type monodentate complexes. We achieved a minimum MAE of 4.0 kcal mol−1 on the monodentate test set, and maintained a comparable MAE of 6.6 kcal mol−1 in the transferability assessment. This approach bypasses the need for multi-reference low-spin optimizations while retaining predictive accuracy, offering a cost-effective strategy for SSE estimation in transition metal chemistry. We hope the insights covered in this study will contribute to the development of additional electronic structure-based descriptors for SSE predictions.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


