基于图转换器的生成对抗网络的候选药物分子靶向从头设计
IF 23.9
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
摘要
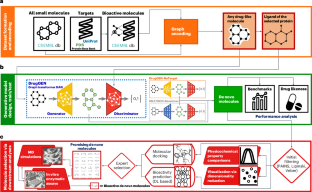
发现新的候选药物分子是药物开发的基本步骤。生成式深度学习模型可以从学习到的概率分布中采样新的分子结构;然而,它们在药物发现中的实际应用取决于生成针对特定目标分子的化合物。在这里,我们介绍DrugGEN,一个端到端生成系统,用于与选定蛋白质相互作用的候选药物分子的从头设计。该方法将分子表示为图形,并使用由图形转换层组成的生成对抗网络对其进行处理。通过对药物样化合物和靶向特异性生物活性分子的大量数据集的训练,DrugGEN设计了AKT1(一种对许多癌症至关重要的激酶)的候选抑制剂。对接和分子动力学模拟表明,生成的化合物有效地与AKT1结合,注意图为模型的推理提供了见解。此外,选择的新生分子被合成,并在体外酶分析的背景下显示出在低微摩尔浓度下抑制AKT1。这些结果证明了DrugGEN在设计靶向分子方面的潜力。使用开放获取的DrugGEN代码库,研究人员可以为其他可药物蛋白质重新训练模型,前提是提供已知生物活性分子的数据集。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Target-specific de novo design of drug candidate molecules with graph-transformer-based generative adversarial networks
Discovering novel drug candidate molecules is a fundamental step in drug development. Generative deep learning models can sample new molecular structures from learned probability distributions; however, their practical use in drug discovery hinges on generating compounds tailored to a specific target molecule. Here we introduce DrugGEN, an end-to-end generative system for the de novo design of drug candidate molecules that interact with a selected protein. The proposed method represents molecules as graphs and processes them using a generative adversarial network that comprises graph transformer layers. Trained on large datasets of drug-like compounds and target-specific bioactive molecules, DrugGEN designed candidate inhibitors for AKT1, a kinase crucial in many cancers. Docking and molecular dynamics simulations suggest that the generated compounds effectively bind to AKT1, and attention maps provide insights into the model’s reasoning. Furthermore, selected de novo molecules were synthesized and shown to inhibit AKT1 at low micromolar concentrations in the context of in vitro enzymatic assays. These results demonstrate the potential of DrugGEN for designing target-specific molecules. Using the open-access DrugGEN codebase, researchers can retrain the model for other druggable proteins, provided a dataset of known bioactive molecules is available. Inhibiting AKT1 kinase can have potentially positive uses against many types of cancer. To find novel molecules targeting this protein, a graph adversarial network is trained as a generative model.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


