{"title":"尿酸钠和骨质疏松症会导致慢性背痛。","authors":"Anna Riegler, Gurpreet Anand","doi":"10.1530/EDM-25-0071","DOIUrl":null,"url":null,"abstract":"<p><strong>Summary: </strong>Alkaptonuria is a rare autosomal recessive metabolic disorder caused by a deficiency in homogentisate 1,2-dioxygenase (HGD), leading to the accumulation of homogentisic acid (HGA) in connective tissues, cartilage, and bones. This accumulation results in multisystem involvement, including early-onset spondyloarthropathy. We present a 54-year-old female from South Tyrol with chronic back and knee pain, accompanied by typical signs of alkaptonuria: ochronosis and darkening of the urine. Molecular genetic testing confirmed the diagnosis of alkaptonuria and identified a previously unreported mutation. Following treatment with nitisinone, a protein-restricted diet, and therapy for osteoporosis, the patient showed significant improvement in symptoms. This case underscores the need to consider rare metabolic disorders in the differential diagnosis of chronic musculoskeletal pain and highlights the importance of early diagnosis and intervention for effective management.</p><p><strong>Learning points: </strong>In cases of early or unexplained degenerative spinal and joint changes in younger individuals, consider secondary metabolic causes. In the presence of the symptom triad - ochronosis, dark urine, and arthropathy - alkaptonuria should be suspected. Alkaptonuria is caused by a rare autosomal recessive defect in homogentisate 1,2-dioxygenase, leading to accumulation of homogentisic acid, which primarily results in the destruction of joints and heart valves. Diagnosis is established through biochemical testing and molecular genetic analysis of the HGD gene. Therapeutic options now include nitisinone as a causal treatment (available since 2020); however, due to often delayed diagnosis, symptomatic management and treatment of sequelae (pain control, joint care, and osteoporosis therapy) continue to play a major role.</p>","PeriodicalId":37467,"journal":{"name":"Endocrinology, Diabetes and Metabolism Case Reports","volume":"2025 3","pages":""},"PeriodicalIF":0.7000,"publicationDate":"2025-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12435171/pdf/","citationCount":"0","resultStr":"{\"title\":\"Combined alkaptonuria and osteoporosis contributing to chronic back pain.\",\"authors\":\"Anna Riegler, Gurpreet Anand\",\"doi\":\"10.1530/EDM-25-0071\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Summary: </strong>Alkaptonuria is a rare autosomal recessive metabolic disorder caused by a deficiency in homogentisate 1,2-dioxygenase (HGD), leading to the accumulation of homogentisic acid (HGA) in connective tissues, cartilage, and bones. This accumulation results in multisystem involvement, including early-onset spondyloarthropathy. We present a 54-year-old female from South Tyrol with chronic back and knee pain, accompanied by typical signs of alkaptonuria: ochronosis and darkening of the urine. Molecular genetic testing confirmed the diagnosis of alkaptonuria and identified a previously unreported mutation. Following treatment with nitisinone, a protein-restricted diet, and therapy for osteoporosis, the patient showed significant improvement in symptoms. This case underscores the need to consider rare metabolic disorders in the differential diagnosis of chronic musculoskeletal pain and highlights the importance of early diagnosis and intervention for effective management.</p><p><strong>Learning points: </strong>In cases of early or unexplained degenerative spinal and joint changes in younger individuals, consider secondary metabolic causes. In the presence of the symptom triad - ochronosis, dark urine, and arthropathy - alkaptonuria should be suspected. Alkaptonuria is caused by a rare autosomal recessive defect in homogentisate 1,2-dioxygenase, leading to accumulation of homogentisic acid, which primarily results in the destruction of joints and heart valves. Diagnosis is established through biochemical testing and molecular genetic analysis of the HGD gene. Therapeutic options now include nitisinone as a causal treatment (available since 2020); however, due to often delayed diagnosis, symptomatic management and treatment of sequelae (pain control, joint care, and osteoporosis therapy) continue to play a major role.</p>\",\"PeriodicalId\":37467,\"journal\":{\"name\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"volume\":\"2025 3\",\"pages\":\"\"},\"PeriodicalIF\":0.7000,\"publicationDate\":\"2025-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12435171/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Endocrinology, Diabetes and Metabolism Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1530/EDM-25-0071\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/1 0:00:00\",\"PubModel\":\"Print\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrinology, Diabetes and Metabolism Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1530/EDM-25-0071","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/1 0:00:00","PubModel":"Print","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Combined alkaptonuria and osteoporosis contributing to chronic back pain.
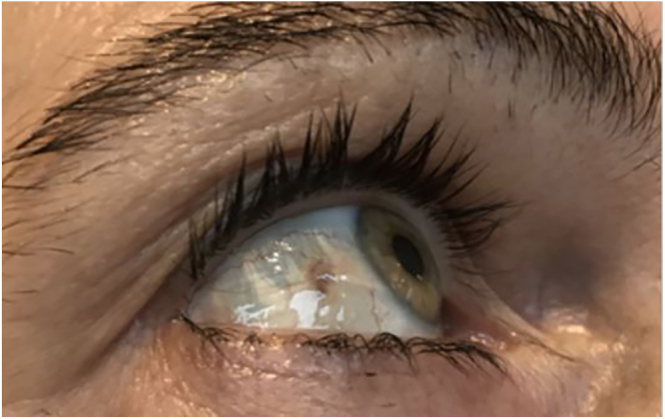
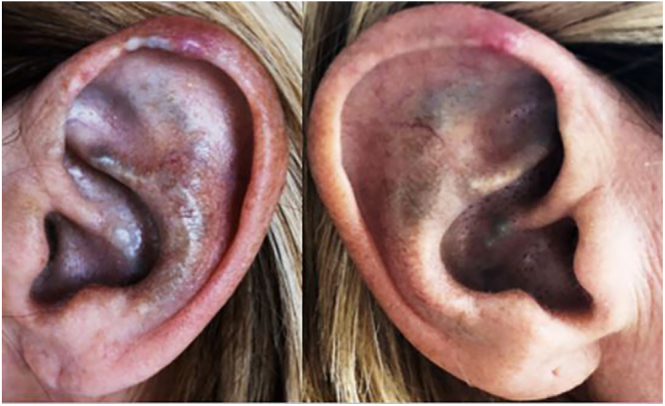
Summary: Alkaptonuria is a rare autosomal recessive metabolic disorder caused by a deficiency in homogentisate 1,2-dioxygenase (HGD), leading to the accumulation of homogentisic acid (HGA) in connective tissues, cartilage, and bones. This accumulation results in multisystem involvement, including early-onset spondyloarthropathy. We present a 54-year-old female from South Tyrol with chronic back and knee pain, accompanied by typical signs of alkaptonuria: ochronosis and darkening of the urine. Molecular genetic testing confirmed the diagnosis of alkaptonuria and identified a previously unreported mutation. Following treatment with nitisinone, a protein-restricted diet, and therapy for osteoporosis, the patient showed significant improvement in symptoms. This case underscores the need to consider rare metabolic disorders in the differential diagnosis of chronic musculoskeletal pain and highlights the importance of early diagnosis and intervention for effective management.
Learning points: In cases of early or unexplained degenerative spinal and joint changes in younger individuals, consider secondary metabolic causes. In the presence of the symptom triad - ochronosis, dark urine, and arthropathy - alkaptonuria should be suspected. Alkaptonuria is caused by a rare autosomal recessive defect in homogentisate 1,2-dioxygenase, leading to accumulation of homogentisic acid, which primarily results in the destruction of joints and heart valves. Diagnosis is established through biochemical testing and molecular genetic analysis of the HGD gene. Therapeutic options now include nitisinone as a causal treatment (available since 2020); however, due to often delayed diagnosis, symptomatic management and treatment of sequelae (pain control, joint care, and osteoporosis therapy) continue to play a major role.
期刊介绍:
Endocrinology, Diabetes & Metabolism Case Reports publishes case reports on common and rare conditions in all areas of clinical endocrinology, diabetes and metabolism. Articles should include clear learning points which readers can use to inform medical education or clinical practice. The types of cases of interest to Endocrinology, Diabetes & Metabolism Case Reports include: -Insight into disease pathogenesis or mechanism of therapy - Novel diagnostic procedure - Novel treatment - Unique/unexpected symptoms or presentations of a disease - New disease or syndrome: presentations/diagnosis/management - Unusual effects of medical treatment - Error in diagnosis/pitfalls and caveats


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


