使用热力学积分优化自由能计算的实用指南
IF 3.1
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
自由能计算在基于结构的药物设计中很有价值,但其准确性和可靠性仍然存在挑战。我们提出了一个基于AMBER20、alchemlyb和开源周期闭合算法的蛋白质-配体结合亲和力估计自动化工作流。通过对四个数据集的178次扰动进行评估,短亚纳秒模拟的效果与MCL1、BACE和CDK2数据集的先前研究相当或更好,而TYK2数据集需要更长的平衡时间(~ 2ns)。|ΔΔG|>;2.0 kcal/mol摄动误差较大,表明这种摄动是不可靠的,从而为改进热力学积分模拟提供了实用的指导。本文章由计算机程序翻译,如有差异,请以英文原文为准。
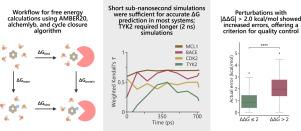
Practical guidelines for optimising free energy calculations using thermodynamic integration
Free energy calculations are valuable in structure-based drug design, but their accuracy and reliability remain challenges. We present an automated workflow for estimation of protein–ligand binding affinity built with AMBER20, alchemlyb, and open-source cycle closure algorithm. Evaluated on 178 perturbations across four datasets, the short sub-nanosecond simulations performed comparably or better than prior studies for the MCL1, BACE, and CDK2 datasets, while the TYK2 dataset required a longer equilibration time (). Perturbations with kcal/mol exhibited higher errors, suggesting such perturbations are unreliable, hence providing a practical guideline for improving thermodynamic integration simulations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Physics Letters
化学-物理:原子、分子和化学物理
CiteScore
5.70
自引率
3.60%
发文量
798
审稿时长
33 days
期刊介绍:
Chemical Physics Letters has an open access mirror journal, Chemical Physics Letters: X, sharing the same aims and scope, editorial team, submission system and rigorous peer review.
Chemical Physics Letters publishes brief reports on molecules, interfaces, condensed phases, nanomaterials and nanostructures, polymers, biomolecular systems, and energy conversion and storage.
Criteria for publication are quality, urgency and impact. Further, experimental results reported in the journal have direct relevance for theory, and theoretical developments or non-routine computations relate directly to experiment. Manuscripts must satisfy these criteria and should not be minor extensions of previous work.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


