以结构为基础的替若替尼衍生物作为细菌色氨酸- trna合成酶抑制剂的设计
IF 4.7
2区 医学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
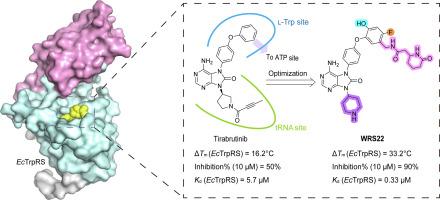
不断升级的抗生素耐药性威胁对全球公共卫生构成了严峻挑战,迫切需要开发具有独特作用机制和独特结构支架的新型治疗剂。色氨酸- trna合成酶(TrpRS)已成为一种很有前景的抗菌靶点。我们前期的研究表明,临床使用的布鲁顿酪氨酸激酶(BTK)抑制剂tirabrutinib及其几种类似物可以同时占据大肠杆菌色氨酸- trna合成酶(EcTrpRS)的底物l-Trp和tRNATrp A76结合位点,从而有效抑制其催化活性。基于这一发现,我们采用基于结构的药物设计方法系统优化了tirabrutinib类似物与l-Trp和tRNATrp结合位点的相互作用,并进一步将结构延伸到ectrpr催化口袋内邻近的ATP结合位点以建立额外的相互作用,从而设计和合成了22个新的衍生物。其中,WRS22(外消旋混合物)与ectrpr结合效果最好,在10 μM浓度下,其ΔTm值为33.2℃,抑制率为90%。其对ectrpr的结合亲和力(Kd = 0.33±0.03 μM)优于阳性对照吲哚霉素(Kd = 0.71±0.1 μM)。值得注意的是,WRS22对人类细胞质trpr (hctrpr)没有亲和力,其与人类BTK的相互作用可能被破坏,表明其具有高度的靶标选择性。因此,结构导向设计成功开发了新的tirabrutinib类似物作为细菌trpr抑制剂,为开发基于aars的抗菌剂提供了一个有前景的先导化合物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Structure-based design of tirabrutinib derivatives as inhibitors of bacterial tryptophanyl-tRNA synthetase
The escalating threat of antibiotic resistance poses a critical challenge to global public health, necessitating the urgent development of novel therapeutic agents with distinct mechanisms of action and unique structural scaffolds. Tryptophanyl-tRNA synthetase (TrpRS) has emerged as a promising antibacterial target. Our previous study demonstrated that the clinically utilized Bruton's tyrosine kinase (BTK) inhibitor tirabrutinib, along with several of its analogues, can simultaneously occupy both the substrates l-Trp and tRNATrp A76 binding sites of Escherichia coli tryptophanyl-tRNA synthetase (EcTrpRS), thereby effectively inhibiting its catalytic activity. Building on this finding, we employed structure-based drug design to systematically optimize the interactions of tirabrutinib analogues with the l-Trp and tRNATrp binding sites, as well as to further extend the structure to the adjacent ATP binding site within the catalytic pocket of EcTrpRS to establish additional interactions, leading to the design and synthesis of 22 new derivatives. Among these, WRS22 (a racemic mixture) demonstrated the best binding to EcTrpRS, with a ΔTm value of 33.2 °C and 90 % inhibition rate at 10 μM concentration. Its binding affinity for EcTrpRS (Kd = 0.33 ± 0.03 μM) is superior to that of the positive control, indolmycin (Kd = 0.71 ± 0.1 μM). Notably, WRS22 displayed no affinity to human cytoplasmic TrpRS (HcTrpRS) and its interaction with human BTK is likely to be disrupted, indicating high degree of target selectivity. Therefore, the structure-guided design successfully developed new tirabrutinib analogues as inhibitors of bacterial TrpRS, presenting a promising lead compound for the development of AARS-based antibacterial agents.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Bioorganic Chemistry
生物-生化与分子生物学
CiteScore
9.70
自引率
3.90%
发文量
679
审稿时长
31 days
期刊介绍:
Bioorganic Chemistry publishes research that addresses biological questions at the molecular level, using organic chemistry and principles of physical organic chemistry. The scope of the journal covers a range of topics at the organic chemistry-biology interface, including: enzyme catalysis, biotransformation and enzyme inhibition; nucleic acids chemistry; medicinal chemistry; natural product chemistry, natural product synthesis and natural product biosynthesis; antimicrobial agents; lipid and peptide chemistry; biophysical chemistry; biological probes; bio-orthogonal chemistry and biomimetic chemistry.
For manuscripts dealing with synthetic bioactive compounds, the Journal requires that the molecular target of the compounds described must be known, and must be demonstrated experimentally in the manuscript. For studies involving natural products, if the molecular target is unknown, some data beyond simple cell-based toxicity studies to provide insight into the mechanism of action is required. Studies supported by molecular docking are welcome, but must be supported by experimental data. The Journal does not consider manuscripts that are purely theoretical or computational in nature.
The Journal publishes regular articles, short communications and reviews. Reviews are normally invited by Editors or Editorial Board members. Authors of unsolicited reviews should first contact an Editor or Editorial Board member to determine whether the proposed article is within the scope of the Journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


