氧化还原可切割二硫连接体通过前药型双功能细胞穿透肽增强siRNA递送
IF 3
3区 医学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
在前药策略中,引入可切割的连接体可以在靶组织内激活。我们开发了一种新的前药型细胞穿透肽(CPP), pi - linkker - crgd,包含一个用于细胞内siRNA递送的立体精细氧化还原可切割连接体。二硫键在基于cpp的前药设计中是有价值的,因为它们在癌细胞的还原环境中是可切割的。我们之前设计了一种双功能CPP, PI-cRGD,通过二硫键将含有α-氨基异丁酸(Aib)的两亲螺旋肽(PI)偶联到αvβ3整合素靶向配体cRGD上。PI-cRGD/siRNA复合物表现出低细胞毒性和通过内吞作用显著的细胞摄取。然而,它未能实现有效的胞质siRNA递送,可能是由于空间位阻阻止了还原细胞表面和细胞内环境中的二硫化物裂解,从而抑制了膜渗透性PI的释放。为了解决这一问题,我们证明了在PI和cRGD之间插入一个立体精细的氧化还原可切割连接体可以保持低毒性和靶向能力,同时可以通过细胞表面的硫醇-二硫交换来切割二硫键,从而激活CPP,促进膜转运,最终实现高效的细胞质siRNA递送。这种简单的可切割连接体策略对各种治疗剂具有广泛的适用性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
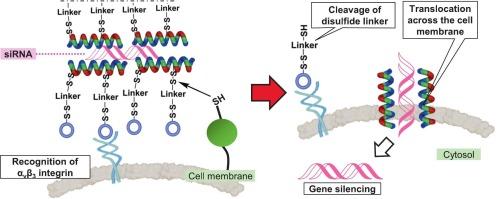
Redox-cleavable disulfide linker enhances siRNA delivery by prodrug-type bifunctional cell-penetrating peptide
In prodrug strategies, introducing a cleavable linker enables activation within target tissues. We developed a novel prodrug-type cell-penetrating peptide (CPP), PI-linker-cRGD, incorporating a sterically refined redox-cleavable linker for intracellular siRNA delivery. Disulfide bonds are valuable in CPP-based prodrug designs because they are cleavable in the reductive environment of cancer cells. We previously designed a bifunctional CPP, PI-cRGD, by conjugating the membrane-permeable α-aminoisobutyric acid (Aib)-containing amphipathic helical peptide (PI) to the αvβ3 integrin-targeting ligand cRGD via a disulfide bond. The PI-cRGD/siRNA complex exhibited low cytotoxicity and significant cellular uptake via endocytosis. However, it failed to achieve efficient cytosolic siRNA delivery, likely due to steric hindrance that prevented the disulfide cleavage in the reductive cell surface and intracellular environments, which in turn inhibited the release of membrane-permeable PI. To address this issue, we demonstrate that inserting a sterically refined redox-cleavable linker between PI and cRGD sustains low toxicity and targeting ability while enabling cleavage of the disulfide bond through thiol–disulfide exchange at the cell surface, thereby activating the CPP, promoting membrane translocation, and ultimately achieving efficient cytosolic siRNA delivery. This simple cleavable-linker strategy holds broad applicability for diverse therapeutic agents.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Bioorganic & Medicinal Chemistry
医学-生化与分子生物学
CiteScore
6.80
自引率
2.90%
发文量
413
审稿时长
17 days
期刊介绍:
Bioorganic & Medicinal Chemistry provides an international forum for the publication of full original research papers and critical reviews on molecular interactions in key biological targets such as receptors, channels, enzymes, nucleotides, lipids and saccharides.
The aim of the journal is to promote a better understanding at the molecular level of life processes, and living organisms, as well as the interaction of these with chemical agents. A special feature will be that colour illustrations will be reproduced at no charge to the author, provided that the Editor agrees that colour is essential to the information content of the illustration in question.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


