含有吲哚基帽基的新型组蛋白去乙酰化酶6抑制剂的设计、合成和生物学评价
IF 2.2
4区 医学
Q3 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
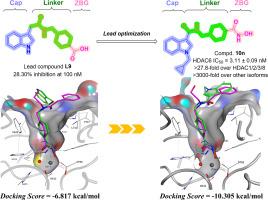
基于我们之前的报道,本研究系统地阐明了一类含有吲哚基帽基的新型HDAC6抑制剂的开发过程。从先导化合物L9开始,通过内部化合物库筛选,进行了系统的结构优化,并设计和合成了一系列衍生物。构效关系(SAR)研究表明,由吲哚环衍生的帽基具有优势,并对不同修饰位点具有偏好。我们最终确定了在吲哚环N-1位置被环丙基甲基取代的化合物10n是最有效的HDAC6抑制剂(IC50 = 3.11±0.09 nM),对HDAC1/2/3/8的选择性比为27.8 ~ 622.2倍,对其他同工异构体的选择性比为3000倍。体外实验进一步证实其对胃癌具有潜在的抗增殖和诱导细胞凋亡的作用,并具有良好的体内药代动力学特性。对接分析显示其与HDAC6催化口袋具有很强的结合亲和力:包括构象的空间互补以及与附近氨基酸残基形成强相互作用。这些发现突出了10n作为开发hdac6选择性抑制剂的有前途的支架,其结构见解指导了未来的优化。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Design, synthesis and biological evaluation of novel histone deacetylase 6 inhibitors containing indole-based cap groups
Based on our previous report, this study systematically elucidated the development process of a novel class of HDAC6 inhibitors containing indole-based cap groups. Starting from lead compound L9 identified via an in-house compound library screening, systematic structural optimization was performed, and a series of derivatives were designed and synthesized. Structure-activity relationship (SAR) studies demonstrated the advantages of cap groups derived from indole ring and the preference of different modification sites. We finally identified compound 10n, substituted with a cyclopropyl-methyl group in the N-1 position of its indole ring, as the most potent HDAC6 inhibitor (IC50 = 3.11 ± 0.09 nM), with selectivity ratios of 27.8- to 622.2-fold over HDAC1/2/3/8 and >3000-fold over other isoforms. In vitro evaluations further demonstrated its potential anti-proliferative and apoptosis-inducing ability in gastric cancer, as well as good in vivo pharmacokinetic properties. Docking analysis revealed its strong binding affinity to the HDAC6 catalytic pocket: including spatial complementarity in conformation and the formation of strong interactions with nearby amino acid residues. These findings highlight 10n as a promising scaffold for developing HDAC6-selective inhibitors, with structural insights guiding future optimizations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
5.70
自引率
3.70%
发文量
463
审稿时长
27 days
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


