共价三嗪框架(CTF-0)作为抗癌药物巯基嘌呤和硫替帕的给药系统:药物吸附的DFT和MD模拟研究
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
本研究利用密度泛函数理论(DFT)和分子动力学(MD)模拟探讨了CTF-0作为抗癌药物巯基嘌呤(MP)和硫替帕(TEP)纳米载体的潜力。MP的- 21.26 kcal/mol和TEP的-22.28 kcal/mol的吸附能表明非共价稳定,主要是通过范德华相互作用,QTAIM和基于IGM的NCI分析证实了这一点。EDA结果表明,静电相互作用是结合的主要稳定因素。FMO分析表明,MP@CTF-0的HOMO-LUMO间隙减小,电荷转移和偶极矩增大,表明溶剂的极性和相互作用增强。100 ns MD仿真进一步证实了MP@CTF-0的结构稳定性。这些发现突出了CTF-0在靶向药物递送应用中作为巯基嘌呤有效载体的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
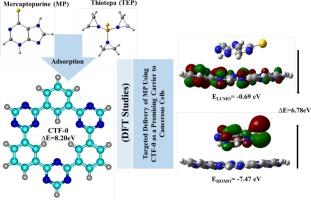
Covalent triazine framework (CTF-0) as a drug delivery system for anti-cancer drugs mercaptopurine and thiotepa: A DFT and MD simulation study on drug adsorption
This study explores the potential of CTF-0 as a nanocarrier for the anticancer drugs Mercaptopurine (MP) and Thiotepa (TEP) using density functional theory (DFT) and molecular dynamics (MD) simulations. Adsorption energies of −21.26 kcal/mol for MP and -22.28 kcal/mol for TEP indicate non-covalent stabilization, primarily through van der Waals interactions as confirmed by QTAIM and IGM based NCI analyses. EDA results reveal that electrostatic interactions are the major stabilizing contributors to binding. FMO analysis shows a reduced HOMO–LUMO gap in MP@CTF-0, supported by greater charge transfer and dipole moment, suggesting enhanced polarity and interaction in solvent. A 100 ns MD simulation further confirms the structural stability of MP@CTF-0. These findings highlight CTF-0's potential as an efficient carrier for Mercaptopurine in targeted drug delivery applications.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


