Natalie A Kukulka, Shriya Singh, Matthew T Whitehead, William B Dobyns, Taeun Chang, Youssef A Kousa
{"title":"桥小脑发育不全:1912 - 2022年回顾。","authors":"Natalie A Kukulka, Shriya Singh, Matthew T Whitehead, William B Dobyns, Taeun Chang, Youssef A Kousa","doi":"10.1093/braincomms/fcaf298","DOIUrl":null,"url":null,"abstract":"<p><p>Pontocerebellar hypoplasia is a rare neurodevelopmental disorder that results from differences in formation and function of the pons, cerebellum and cerebrum. It can be diagnosed prenatally or postnatally with a combination of clinical, neuroimaging and genetic data obtained over time. The diagnosis of pontocerebellar hypoplasia usually portends severe developmental delay, epilepsy and/or neurodegeneration in childhood. Here we perform a comprehensive review with the primary goal of evaluating published evidence addressing the clinical and genetic features of pontocerebellar hypoplasia by type and subtype. Secondly, we summarize neurodiagnostic patterns of pontocerebellar hypoplasia and demonstrate its spectrum. Finally, we provide recommendations in diagnosis, prognosis and management for the neurologist. To address these goals, we performed an extensive review of published literature from 1912 to 2022. We identified 191 publications by combining search results from PubMed, OMIM and cross-referenced bibliographies. Publications on developmental neuroanatomy, not pertaining to pontocerebellar hypoplasia or published in a foreign language were excluded. We performed both qualitative (1912-1993) and quantitative (1993-2022) analyses to understand the current classification of this disease as it pertains to genetic and neurodiagnostic features of pontocerebellar hypoplasia by type and subtype. Our review shows that the most reported types of pontocerebellar hypoplasia are 1, 2 and 6; less frequently described are 3, 4 and 9. Very few cases are described for all other subsequent pontocerebellar hypoplasia types. Mutations in <i>TSEN54</i>, <i>RARS2</i>, <i>EXOSC3</i> and <i>AMPD2</i> (genes that regulate RNA processing and basic cellular metabolism) are the most frequently reported pathological mutations in pontocerebellar hypoplasia. The neuroradiographic features of pontocerebellar hypoplasia are complex and evolve over time, affecting the pons, cerebellum, vermis, cortex and cerebral white matter. In conclusion, pontocerebellar hypoplasia is a rare neurodevelopmental disorder, often the result of genetic dysfunction in basic neural metabolism. The diagnosis conveys significant implications for the affected individual and their families and requires a combination of clinical, neuroradiographic, and genetic testing to best inform type/subtype categorization of pontocerebellar hypoplasia.</p>","PeriodicalId":93915,"journal":{"name":"Brain communications","volume":"7 5","pages":"fcaf298"},"PeriodicalIF":4.5000,"publicationDate":"2025-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12422213/pdf/","citationCount":"0","resultStr":"{\"title\":\"Pontocerebellar hypoplasia: a review from 1912 to 2022.\",\"authors\":\"Natalie A Kukulka, Shriya Singh, Matthew T Whitehead, William B Dobyns, Taeun Chang, Youssef A Kousa\",\"doi\":\"10.1093/braincomms/fcaf298\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pontocerebellar hypoplasia is a rare neurodevelopmental disorder that results from differences in formation and function of the pons, cerebellum and cerebrum. It can be diagnosed prenatally or postnatally with a combination of clinical, neuroimaging and genetic data obtained over time. The diagnosis of pontocerebellar hypoplasia usually portends severe developmental delay, epilepsy and/or neurodegeneration in childhood. Here we perform a comprehensive review with the primary goal of evaluating published evidence addressing the clinical and genetic features of pontocerebellar hypoplasia by type and subtype. Secondly, we summarize neurodiagnostic patterns of pontocerebellar hypoplasia and demonstrate its spectrum. Finally, we provide recommendations in diagnosis, prognosis and management for the neurologist. To address these goals, we performed an extensive review of published literature from 1912 to 2022. We identified 191 publications by combining search results from PubMed, OMIM and cross-referenced bibliographies. Publications on developmental neuroanatomy, not pertaining to pontocerebellar hypoplasia or published in a foreign language were excluded. We performed both qualitative (1912-1993) and quantitative (1993-2022) analyses to understand the current classification of this disease as it pertains to genetic and neurodiagnostic features of pontocerebellar hypoplasia by type and subtype. Our review shows that the most reported types of pontocerebellar hypoplasia are 1, 2 and 6; less frequently described are 3, 4 and 9. Very few cases are described for all other subsequent pontocerebellar hypoplasia types. Mutations in <i>TSEN54</i>, <i>RARS2</i>, <i>EXOSC3</i> and <i>AMPD2</i> (genes that regulate RNA processing and basic cellular metabolism) are the most frequently reported pathological mutations in pontocerebellar hypoplasia. The neuroradiographic features of pontocerebellar hypoplasia are complex and evolve over time, affecting the pons, cerebellum, vermis, cortex and cerebral white matter. In conclusion, pontocerebellar hypoplasia is a rare neurodevelopmental disorder, often the result of genetic dysfunction in basic neural metabolism. The diagnosis conveys significant implications for the affected individual and their families and requires a combination of clinical, neuroradiographic, and genetic testing to best inform type/subtype categorization of pontocerebellar hypoplasia.</p>\",\"PeriodicalId\":93915,\"journal\":{\"name\":\"Brain communications\",\"volume\":\"7 5\",\"pages\":\"fcaf298\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2025-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12422213/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Brain communications\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/braincomms/fcaf298\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/braincomms/fcaf298","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Pontocerebellar hypoplasia: a review from 1912 to 2022.
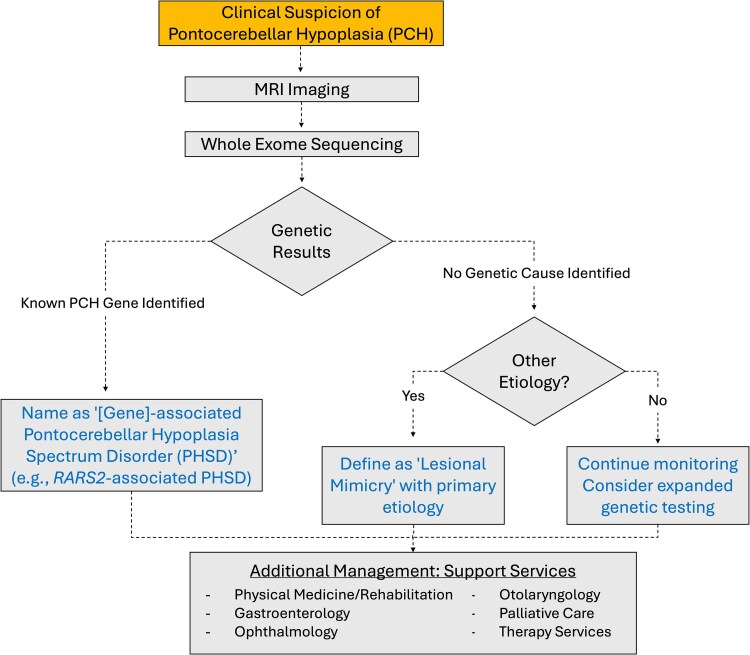
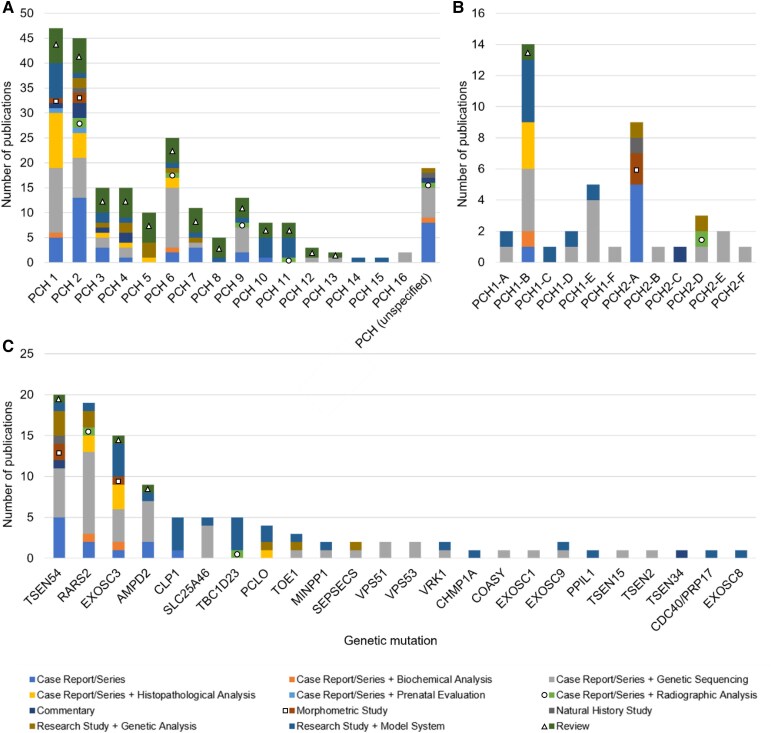
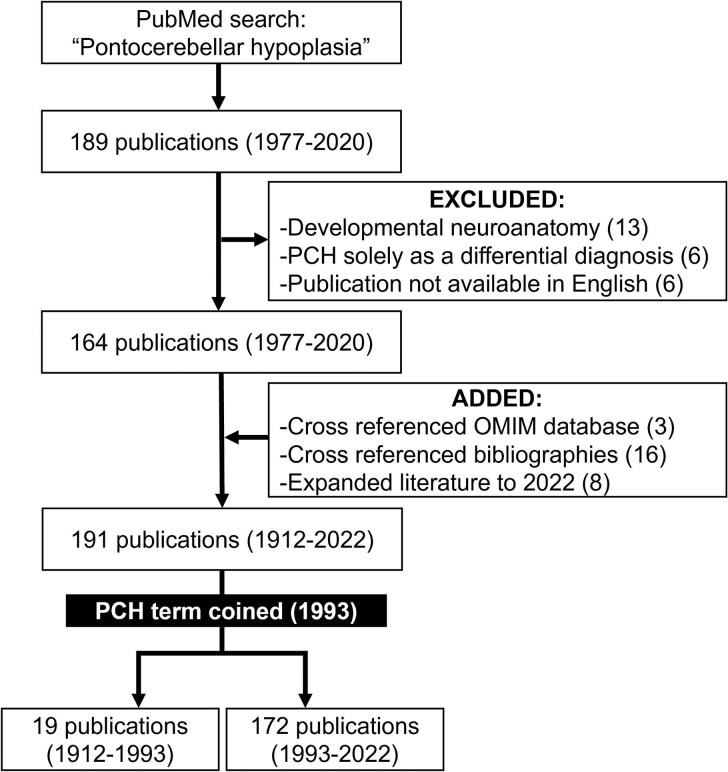
Pontocerebellar hypoplasia is a rare neurodevelopmental disorder that results from differences in formation and function of the pons, cerebellum and cerebrum. It can be diagnosed prenatally or postnatally with a combination of clinical, neuroimaging and genetic data obtained over time. The diagnosis of pontocerebellar hypoplasia usually portends severe developmental delay, epilepsy and/or neurodegeneration in childhood. Here we perform a comprehensive review with the primary goal of evaluating published evidence addressing the clinical and genetic features of pontocerebellar hypoplasia by type and subtype. Secondly, we summarize neurodiagnostic patterns of pontocerebellar hypoplasia and demonstrate its spectrum. Finally, we provide recommendations in diagnosis, prognosis and management for the neurologist. To address these goals, we performed an extensive review of published literature from 1912 to 2022. We identified 191 publications by combining search results from PubMed, OMIM and cross-referenced bibliographies. Publications on developmental neuroanatomy, not pertaining to pontocerebellar hypoplasia or published in a foreign language were excluded. We performed both qualitative (1912-1993) and quantitative (1993-2022) analyses to understand the current classification of this disease as it pertains to genetic and neurodiagnostic features of pontocerebellar hypoplasia by type and subtype. Our review shows that the most reported types of pontocerebellar hypoplasia are 1, 2 and 6; less frequently described are 3, 4 and 9. Very few cases are described for all other subsequent pontocerebellar hypoplasia types. Mutations in TSEN54, RARS2, EXOSC3 and AMPD2 (genes that regulate RNA processing and basic cellular metabolism) are the most frequently reported pathological mutations in pontocerebellar hypoplasia. The neuroradiographic features of pontocerebellar hypoplasia are complex and evolve over time, affecting the pons, cerebellum, vermis, cortex and cerebral white matter. In conclusion, pontocerebellar hypoplasia is a rare neurodevelopmental disorder, often the result of genetic dysfunction in basic neural metabolism. The diagnosis conveys significant implications for the affected individual and their families and requires a combination of clinical, neuroradiographic, and genetic testing to best inform type/subtype categorization of pontocerebellar hypoplasia.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


