{"title":"肌酸酐互变异构机理的量子化学计算及化学动力学研究","authors":"Yiqing Sun, Xiankai Jiang, Zhenhai Xiong, Junjian Miao","doi":"10.1007/s00894-025-06501-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Thermal processing of meat typically leads to the formation of heterocyclic aromatic amines (HAAs), a class of highly toxic compounds. Although extensive research has been conducted on HAAs, the precise formation mechanisms of individual HAAs remain incompletely understood, with many critical details yet to be elucidated. Several unresolved aspects concern the number of tautomers of creatinine—the key precursor in HAA formation, the interconversion pathways among these tautomers and the time scales involved, and the equilibrium distribution ratios among the tautomeric forms. Addressing these questions is essential for achieving an accurate understanding of the mechanistic pathways underlying the formation of HAAs.</p><h3>Methods</h3><p>All possible tautomers were manually deduced and verified by RDkit, a chemoinformatics toolkit. Geometry optimizations and frequency analyses were performed using density functional theory (DFT). The distribution of various tautomers was evaluated under three different environments—gas phase, ethanol, and water—to simulate real conditions. Calculations were carried out at the B3LYP/def2QZVPP//6-31G(d,p) level of theory, with the polarizable continuum model (PCM) applied for ethanol and water. A similar computational approach to the calculations on distribution was employed to investigate tautomerization mechanisms. Tautomerization kinetics were analyzed within the framework of transition state theory (TST) to determine rate constants for each tautomeric interconversion. Tunneling correction factors (κ) were then calculated to account for quantum mechanical tunneling effects. Subsequently, the corresponding system of differential equations was solved to obtain the time-dependent concentration profiles of each tautomeric species.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 10","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Quantum chemistry calculations and chemical kinetic studies on the tautomeric mechanism of creatinine\",\"authors\":\"Yiqing Sun, Xiankai Jiang, Zhenhai Xiong, Junjian Miao\",\"doi\":\"10.1007/s00894-025-06501-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>Thermal processing of meat typically leads to the formation of heterocyclic aromatic amines (HAAs), a class of highly toxic compounds. Although extensive research has been conducted on HAAs, the precise formation mechanisms of individual HAAs remain incompletely understood, with many critical details yet to be elucidated. Several unresolved aspects concern the number of tautomers of creatinine—the key precursor in HAA formation, the interconversion pathways among these tautomers and the time scales involved, and the equilibrium distribution ratios among the tautomeric forms. Addressing these questions is essential for achieving an accurate understanding of the mechanistic pathways underlying the formation of HAAs.</p><h3>Methods</h3><p>All possible tautomers were manually deduced and verified by RDkit, a chemoinformatics toolkit. Geometry optimizations and frequency analyses were performed using density functional theory (DFT). The distribution of various tautomers was evaluated under three different environments—gas phase, ethanol, and water—to simulate real conditions. Calculations were carried out at the B3LYP/def2QZVPP//6-31G(d,p) level of theory, with the polarizable continuum model (PCM) applied for ethanol and water. A similar computational approach to the calculations on distribution was employed to investigate tautomerization mechanisms. Tautomerization kinetics were analyzed within the framework of transition state theory (TST) to determine rate constants for each tautomeric interconversion. Tunneling correction factors (κ) were then calculated to account for quantum mechanical tunneling effects. Subsequently, the corresponding system of differential equations was solved to obtain the time-dependent concentration profiles of each tautomeric species.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 10\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-09-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06501-w\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06501-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Quantum chemistry calculations and chemical kinetic studies on the tautomeric mechanism of creatinine
Context
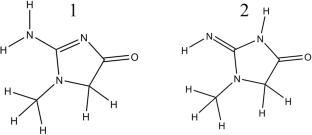
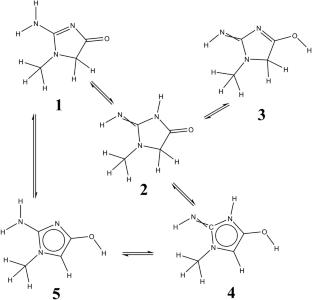
Thermal processing of meat typically leads to the formation of heterocyclic aromatic amines (HAAs), a class of highly toxic compounds. Although extensive research has been conducted on HAAs, the precise formation mechanisms of individual HAAs remain incompletely understood, with many critical details yet to be elucidated. Several unresolved aspects concern the number of tautomers of creatinine—the key precursor in HAA formation, the interconversion pathways among these tautomers and the time scales involved, and the equilibrium distribution ratios among the tautomeric forms. Addressing these questions is essential for achieving an accurate understanding of the mechanistic pathways underlying the formation of HAAs.
Methods
All possible tautomers were manually deduced and verified by RDkit, a chemoinformatics toolkit. Geometry optimizations and frequency analyses were performed using density functional theory (DFT). The distribution of various tautomers was evaluated under three different environments—gas phase, ethanol, and water—to simulate real conditions. Calculations were carried out at the B3LYP/def2QZVPP//6-31G(d,p) level of theory, with the polarizable continuum model (PCM) applied for ethanol and water. A similar computational approach to the calculations on distribution was employed to investigate tautomerization mechanisms. Tautomerization kinetics were analyzed within the framework of transition state theory (TST) to determine rate constants for each tautomeric interconversion. Tunneling correction factors (κ) were then calculated to account for quantum mechanical tunneling effects. Subsequently, the corresponding system of differential equations was solved to obtain the time-dependent concentration profiles of each tautomeric species.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


