结构,电子和热力学表征与光谱,拓扑,反应性,和分子对接研究二烯丙基硫化
IF 3
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
摘要
利用密度泛函理论(DFT)对大蒜和洋葱中的生物活性有机硫化合物二烯丙基硫醚(DAS)进行了分析。DAS具有抗菌和抗癌特性,使其成为药物发现的潜在候选者。几何优化显示键长和键角与电子离域一致。前沿分子轨道分析表明,极性溶剂中HOMO-LUMO间隙增大,稳定性提高。自然键轨道分析证实了通过硫中心相互作用产生的显著电荷离域,解释了部分S-C双键特征和相邻CH键的减弱,从而支持亲电性趋势。在极性溶剂中,由于激发态失稳,紫外-可见光谱显示λmax减小。态密度表示导带附近的多余电子密度。减小的密度梯度图确定了稳定的范德华相互作用。通过福井函数分析验证了亲电(硫)和亲核(烯丙基H)位点的分子静电电位定位。药物相似度评价表明,DAS满足Lipinski和Veber的口服生物利用度规则,但需要改进结构以提高复杂性。与蛋白3IAI、3E4E和4EY7分子对接发现疏水和π -硫相互作用,证实了它们的生物学相关性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
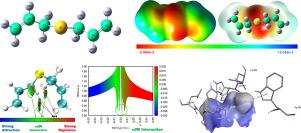
Structural, electronic, and thermodynamic characterization with spectroscopic, topological, reactivity, and molecular docking studies of diallyl sulfide
The bioactive organosulfur compound diallyl sulfide (DAS), found in garlic and onions, was analyzed using density functional theory (DFT). DAS exhibits antimicrobial and anticancer properties, making it a potential candidate for drug discovery. Geometry optimization revealed bond lengths and angles consistent with electron delocalization. Frontier molecular orbital analysis showed increased HOMO–LUMO gaps and stability in polar solvents. Natural bond orbital analysis confirmed significant charge delocalization via sulfur-centered interactions, explaining the partial S–C double-bond character and weakened adjacent C![]() H bonds, thus supporting electrophilicity trends. Thermodynamic properties displayed temperature-dependent changes, while UV–Vis spectra showed reduced in polar solvents owing to excited-state destabilization. The density of states indicates the excess electron density near the conduction band. The reduced density gradient plots identified the stabilizing van der Waals interactions. Molecular electrostatic potential mapping of electrophilic (sulfur) and nucleophilic (allyl H) sites was validated by Fukui function analysis. Druglikeness evaluation indicated that DAS satisfies Lipinski’s and Veber’s rules for oral bioavailability, but requires structural refinement to enhance complexity. Molecular docking with proteins 3IAI, 3E4E, and 4EY7 revealed hydrophobic and –sulfur interactions, confirming their biological relevance.
H bonds, thus supporting electrophilicity trends. Thermodynamic properties displayed temperature-dependent changes, while UV–Vis spectra showed reduced in polar solvents owing to excited-state destabilization. The density of states indicates the excess electron density near the conduction band. The reduced density gradient plots identified the stabilizing van der Waals interactions. Molecular electrostatic potential mapping of electrophilic (sulfur) and nucleophilic (allyl H) sites was validated by Fukui function analysis. Druglikeness evaluation indicated that DAS satisfies Lipinski’s and Veber’s rules for oral bioavailability, but requires structural refinement to enhance complexity. Molecular docking with proteins 3IAI, 3E4E, and 4EY7 revealed hydrophobic and –sulfur interactions, confirming their biological relevance.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


