{"title":"肌强直性营养不良小鼠模型中肌肉驱动的脊髓组织学和转录组学改变:对神经病变的见解。","authors":"Guanzhong Shi, Yining Luan, Yuzhen Ouyang, Kangzhi Chen, Kaiyue Zhang, Zeyi Wen, Huan Yang, Kun Huang","doi":"10.1093/braincomms/fcaf313","DOIUrl":null,"url":null,"abstract":"<p><p>Myotonic dystrophy type 1 (DM1) is an inherited neuromuscular disorder characterized by muscle weakness, atrophy and myotonia, with multi-system involvement. Recent studies have highlighted the pathological heterogeneity within the CNS of DM1 patients, particularly significant changes in spinal transcriptome expression and alternative splicing. In this study, we conducted a comprehensive transcriptome analysis of the spinal cord in the muscle-specific DM1 mouse model and their wild-type controls across different life stages: young, adult and old age. Our results revealed an age-dependent increase in differential gene expression between DM1 and wild-type mice with a predominance of downregulated genes. Notably, five genes (<i>Adgre1</i>, <i>Ccl3</i>, <i>Fcrls</i>, <i>Ogfrl1</i> and <i>Reg3b</i>) were consistently differentially expressed across all age groups. We also generated a temporal profile of cell-type proportions and observed reductions in microglia and astrocytes, along with a trend towards increased ventral neuron populations. Additionally, we characterized the temporal splicing alterations in the spinal cord of DM1 mice and compared with homologous exon skipping events in the CNS of DM1 patients. Our RNA sequencing data elucidate the molecular and cellular adaptations of the spinal cord to muscle defects over time, underscoring that splicing abnormalities observed in the CNS of DM1 patients may reflect contributions from muscle pathology. These findings highlight the necessity of a holistic approach to comprehensively understand the complexity of DM1.</p>","PeriodicalId":93915,"journal":{"name":"Brain communications","volume":"7 5","pages":"fcaf313"},"PeriodicalIF":4.5000,"publicationDate":"2025-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409276/pdf/","citationCount":"0","resultStr":"{\"title\":\"Muscle-driven spinal cord histological and transcriptomic alterations in a myotonic dystrophy mouse model: insights into neuropathy.\",\"authors\":\"Guanzhong Shi, Yining Luan, Yuzhen Ouyang, Kangzhi Chen, Kaiyue Zhang, Zeyi Wen, Huan Yang, Kun Huang\",\"doi\":\"10.1093/braincomms/fcaf313\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Myotonic dystrophy type 1 (DM1) is an inherited neuromuscular disorder characterized by muscle weakness, atrophy and myotonia, with multi-system involvement. Recent studies have highlighted the pathological heterogeneity within the CNS of DM1 patients, particularly significant changes in spinal transcriptome expression and alternative splicing. In this study, we conducted a comprehensive transcriptome analysis of the spinal cord in the muscle-specific DM1 mouse model and their wild-type controls across different life stages: young, adult and old age. Our results revealed an age-dependent increase in differential gene expression between DM1 and wild-type mice with a predominance of downregulated genes. Notably, five genes (<i>Adgre1</i>, <i>Ccl3</i>, <i>Fcrls</i>, <i>Ogfrl1</i> and <i>Reg3b</i>) were consistently differentially expressed across all age groups. We also generated a temporal profile of cell-type proportions and observed reductions in microglia and astrocytes, along with a trend towards increased ventral neuron populations. Additionally, we characterized the temporal splicing alterations in the spinal cord of DM1 mice and compared with homologous exon skipping events in the CNS of DM1 patients. Our RNA sequencing data elucidate the molecular and cellular adaptations of the spinal cord to muscle defects over time, underscoring that splicing abnormalities observed in the CNS of DM1 patients may reflect contributions from muscle pathology. These findings highlight the necessity of a holistic approach to comprehensively understand the complexity of DM1.</p>\",\"PeriodicalId\":93915,\"journal\":{\"name\":\"Brain communications\",\"volume\":\"7 5\",\"pages\":\"fcaf313\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2025-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409276/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Brain communications\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/braincomms/fcaf313\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/braincomms/fcaf313","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Muscle-driven spinal cord histological and transcriptomic alterations in a myotonic dystrophy mouse model: insights into neuropathy.
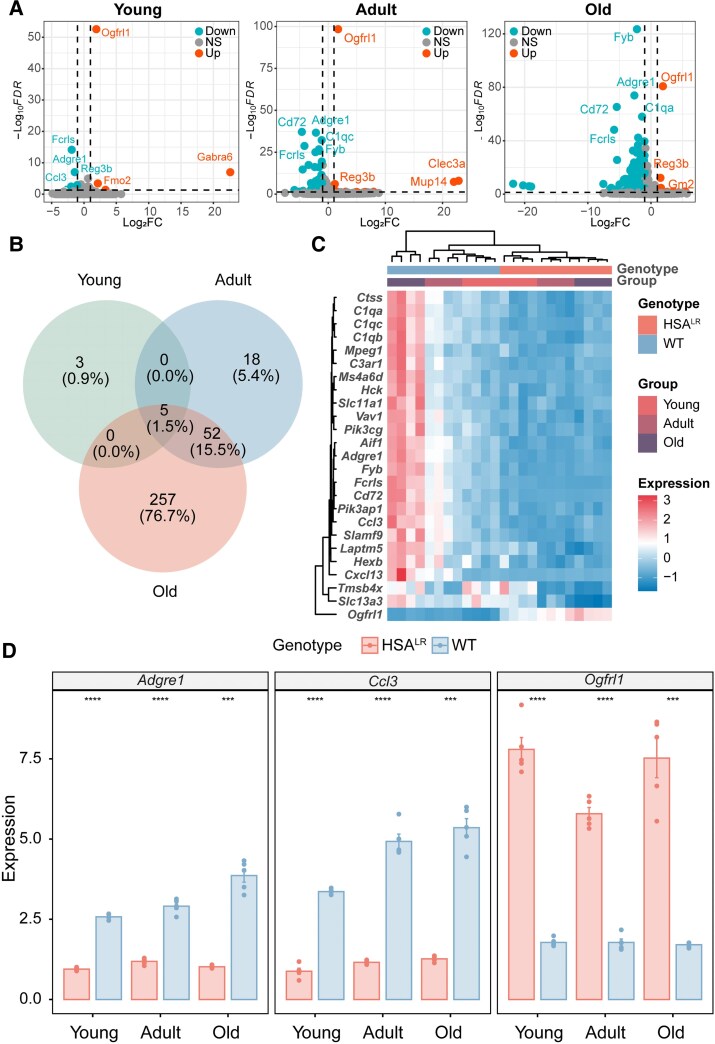
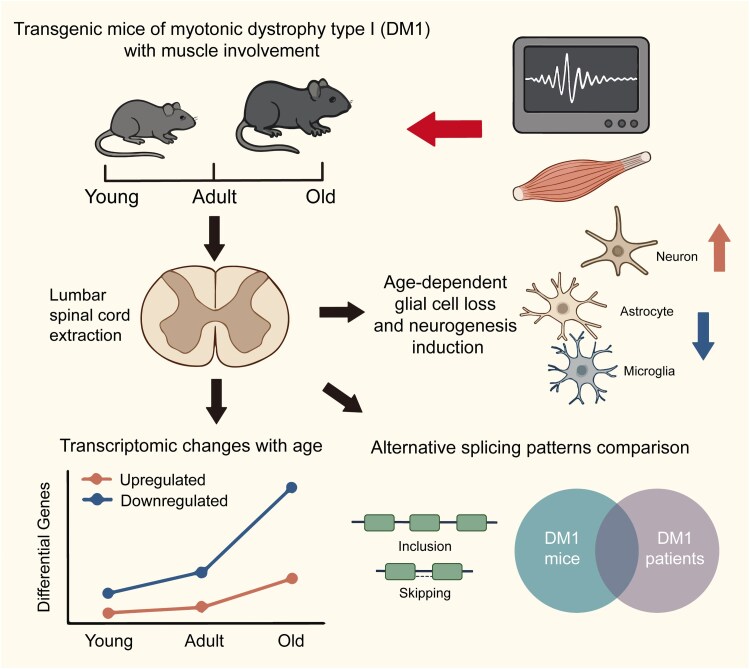
Myotonic dystrophy type 1 (DM1) is an inherited neuromuscular disorder characterized by muscle weakness, atrophy and myotonia, with multi-system involvement. Recent studies have highlighted the pathological heterogeneity within the CNS of DM1 patients, particularly significant changes in spinal transcriptome expression and alternative splicing. In this study, we conducted a comprehensive transcriptome analysis of the spinal cord in the muscle-specific DM1 mouse model and their wild-type controls across different life stages: young, adult and old age. Our results revealed an age-dependent increase in differential gene expression between DM1 and wild-type mice with a predominance of downregulated genes. Notably, five genes (Adgre1, Ccl3, Fcrls, Ogfrl1 and Reg3b) were consistently differentially expressed across all age groups. We also generated a temporal profile of cell-type proportions and observed reductions in microglia and astrocytes, along with a trend towards increased ventral neuron populations. Additionally, we characterized the temporal splicing alterations in the spinal cord of DM1 mice and compared with homologous exon skipping events in the CNS of DM1 patients. Our RNA sequencing data elucidate the molecular and cellular adaptations of the spinal cord to muscle defects over time, underscoring that splicing abnormalities observed in the CNS of DM1 patients may reflect contributions from muscle pathology. These findings highlight the necessity of a holistic approach to comprehensively understand the complexity of DM1.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


