{"title":"比例:一个R包推断基因转录率与新颖的最小平方和方法。","authors":"Yu Liu, Fadhl Alakwaa","doi":"10.1093/nargab/lqaf123","DOIUrl":null,"url":null,"abstract":"<p><p>The dynamics of transcriptional elongation influence many biological activities, such as RNA splicing, polyadenylation, and nuclear export. To quantify the elongation rate, a typical method is to treat cells with drugs that inhibit RNA polymerase II (Pol II) from entering the gene body and then track Pol II using Pro-seq or Gro-seq. However, the downstream data analysis is challenged by the problem of identifying the transition point between the gene regions inhibited by the drug and not, which is necessary to calculate the transcription rate. Although the traditional hidden Markov model (HMM) can be used to solve it, this method is complicated with its hidden variable and many parameters to be estimated. Hence, we developed the R package <i>proRate</i>, which identifies the transition point with a novel least sum of squares (LSS) method and calculates the elongation rate accordingly. In addition, <i>proRate</i> also covers other functions frequently used in transcription dynamic study, including metagene plotting, pause index calculation, gene structure analysis, etc. The effectiveness of this package is proved by its performance on three Pro-seq or Gro-seq datasets, showing higher accuracy than HMM. <i>proRate</i> is freely available at https://github.com/yuabrahamliu/proRate or https://github.com/FADHLyemen/proRate.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"7 3","pages":"lqaf123"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12412782/pdf/","citationCount":"0","resultStr":"{\"title\":\"<i>proRate</i>: an R package to infer gene transcription rates with a novel least sum of squares method.\",\"authors\":\"Yu Liu, Fadhl Alakwaa\",\"doi\":\"10.1093/nargab/lqaf123\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The dynamics of transcriptional elongation influence many biological activities, such as RNA splicing, polyadenylation, and nuclear export. To quantify the elongation rate, a typical method is to treat cells with drugs that inhibit RNA polymerase II (Pol II) from entering the gene body and then track Pol II using Pro-seq or Gro-seq. However, the downstream data analysis is challenged by the problem of identifying the transition point between the gene regions inhibited by the drug and not, which is necessary to calculate the transcription rate. Although the traditional hidden Markov model (HMM) can be used to solve it, this method is complicated with its hidden variable and many parameters to be estimated. Hence, we developed the R package <i>proRate</i>, which identifies the transition point with a novel least sum of squares (LSS) method and calculates the elongation rate accordingly. In addition, <i>proRate</i> also covers other functions frequently used in transcription dynamic study, including metagene plotting, pause index calculation, gene structure analysis, etc. The effectiveness of this package is proved by its performance on three Pro-seq or Gro-seq datasets, showing higher accuracy than HMM. <i>proRate</i> is freely available at https://github.com/yuabrahamliu/proRate or https://github.com/FADHLyemen/proRate.</p>\",\"PeriodicalId\":33994,\"journal\":{\"name\":\"NAR Genomics and Bioinformatics\",\"volume\":\"7 3\",\"pages\":\"lqaf123\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-09-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12412782/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Genomics and Bioinformatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/nargab/lqaf123\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqaf123","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
proRate: an R package to infer gene transcription rates with a novel least sum of squares method.
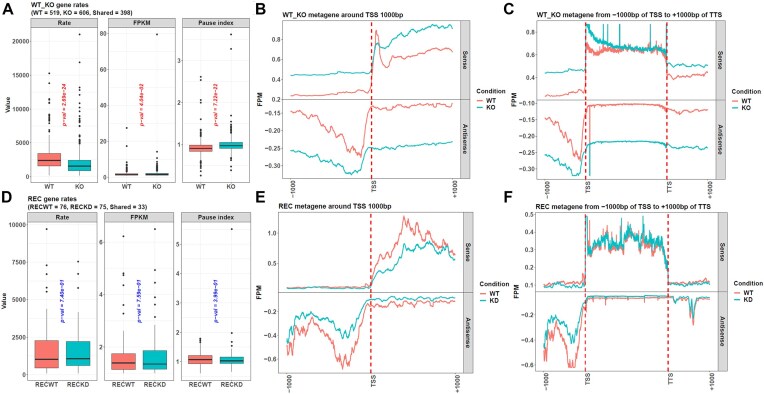
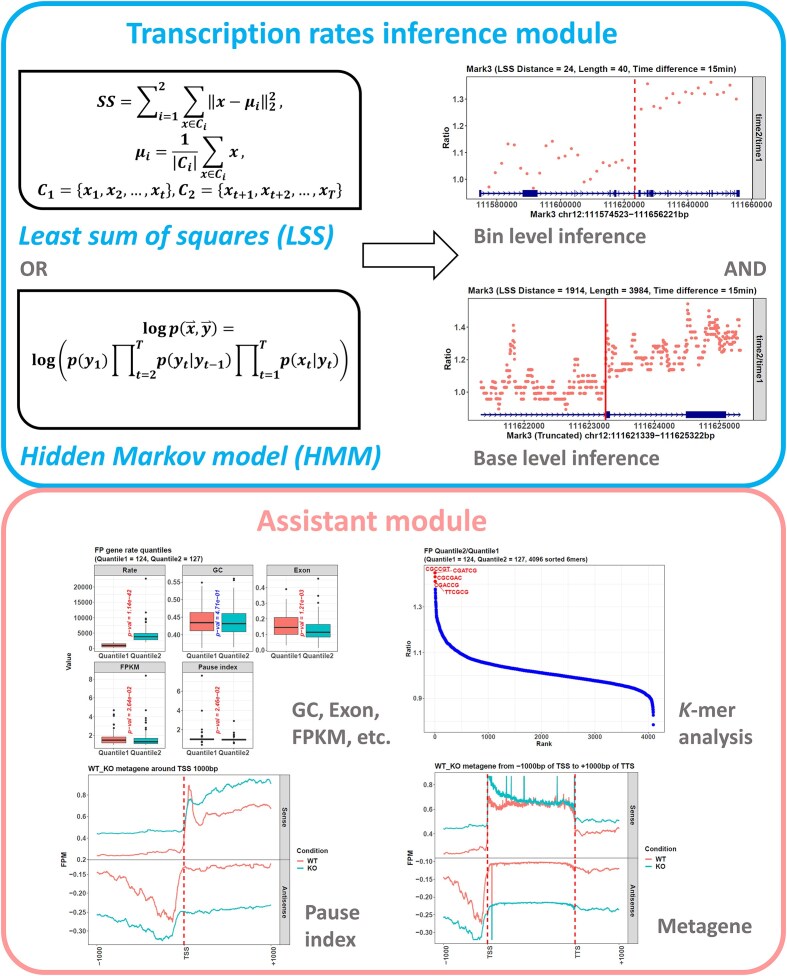
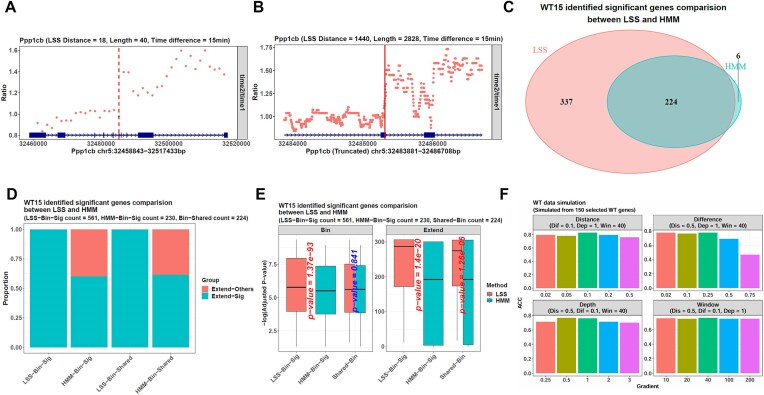
The dynamics of transcriptional elongation influence many biological activities, such as RNA splicing, polyadenylation, and nuclear export. To quantify the elongation rate, a typical method is to treat cells with drugs that inhibit RNA polymerase II (Pol II) from entering the gene body and then track Pol II using Pro-seq or Gro-seq. However, the downstream data analysis is challenged by the problem of identifying the transition point between the gene regions inhibited by the drug and not, which is necessary to calculate the transcription rate. Although the traditional hidden Markov model (HMM) can be used to solve it, this method is complicated with its hidden variable and many parameters to be estimated. Hence, we developed the R package proRate, which identifies the transition point with a novel least sum of squares (LSS) method and calculates the elongation rate accordingly. In addition, proRate also covers other functions frequently used in transcription dynamic study, including metagene plotting, pause index calculation, gene structure analysis, etc. The effectiveness of this package is proved by its performance on three Pro-seq or Gro-seq datasets, showing higher accuracy than HMM. proRate is freely available at https://github.com/yuabrahamliu/proRate or https://github.com/FADHLyemen/proRate.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


