Sara Missaglia, Eleonora Martegani, Corrado Angelini, Rita Horvath, Veronika Karcagi, Endre Pal, Daniela Tavian
{"title":"病例报告:致病性PNPLA2变异和无义介导的mRNA衰变导致早发性中性脂质储存病伴肌病。","authors":"Sara Missaglia, Eleonora Martegani, Corrado Angelini, Rita Horvath, Veronika Karcagi, Endre Pal, Daniela Tavian","doi":"10.3389/fgene.2025.1642442","DOIUrl":null,"url":null,"abstract":"<p><p>Neutral Lipid Storage Disease with Myopathy (NLSDM) is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues. It typically manifests with progressive skeletal myopathy, with an onset of around 35 years. In addition, some patients develop cardiomyopathy and liver dysfunction. Herein, we report the molecular characterization of a 27-year-old Hungarian patient and his family in whom two severe <i>PNPLA2</i> mutations led to complete loss of ATGL production in the patient's tissues. DNA sequencing revealed a nonsense (c.24G>A) and a frameshift mutation (c.798dupC) in the <i>PNPLA2</i> gene. RNA analysis showed nonsense-mediated decay of the c.798dupC transcript, while c.24G>A was normally expressed in the patient. However, Western blot analysis did not detect ATGL protein production. From a clinical perspective, our patient exhibited pes planus, proximal muscle weakness of the lower limbs and elevated CK levels from the age of six. MRI and biopsy revealed lipid accumulation, and leukocytes showed Jordans' anomaly. The muscle weakness progressed, and cardiomyopathy and hepatic steatosis were also observed recently. The case highlights two severe <i>PNPLA2</i> mutations leading to complete ATGL deficiency and an unusual early-onset myopathy in childhood.</p>","PeriodicalId":12750,"journal":{"name":"Frontiers in Genetics","volume":"16 ","pages":"1642442"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12408259/pdf/","citationCount":"0","resultStr":"{\"title\":\"Case Report: Pathogenic <i>PNPLA2</i> variants and nonsense-mediated mRNA decay result in an early-onset neutral lipid storage disease with myopathy.\",\"authors\":\"Sara Missaglia, Eleonora Martegani, Corrado Angelini, Rita Horvath, Veronika Karcagi, Endre Pal, Daniela Tavian\",\"doi\":\"10.3389/fgene.2025.1642442\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neutral Lipid Storage Disease with Myopathy (NLSDM) is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues. It typically manifests with progressive skeletal myopathy, with an onset of around 35 years. In addition, some patients develop cardiomyopathy and liver dysfunction. Herein, we report the molecular characterization of a 27-year-old Hungarian patient and his family in whom two severe <i>PNPLA2</i> mutations led to complete loss of ATGL production in the patient's tissues. DNA sequencing revealed a nonsense (c.24G>A) and a frameshift mutation (c.798dupC) in the <i>PNPLA2</i> gene. RNA analysis showed nonsense-mediated decay of the c.798dupC transcript, while c.24G>A was normally expressed in the patient. However, Western blot analysis did not detect ATGL protein production. From a clinical perspective, our patient exhibited pes planus, proximal muscle weakness of the lower limbs and elevated CK levels from the age of six. MRI and biopsy revealed lipid accumulation, and leukocytes showed Jordans' anomaly. The muscle weakness progressed, and cardiomyopathy and hepatic steatosis were also observed recently. The case highlights two severe <i>PNPLA2</i> mutations leading to complete ATGL deficiency and an unusual early-onset myopathy in childhood.</p>\",\"PeriodicalId\":12750,\"journal\":{\"name\":\"Frontiers in Genetics\",\"volume\":\"16 \",\"pages\":\"1642442\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-08-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12408259/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.3389/fgene.2025.1642442\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.3389/fgene.2025.1642442","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Case Report: Pathogenic PNPLA2 variants and nonsense-mediated mRNA decay result in an early-onset neutral lipid storage disease with myopathy.
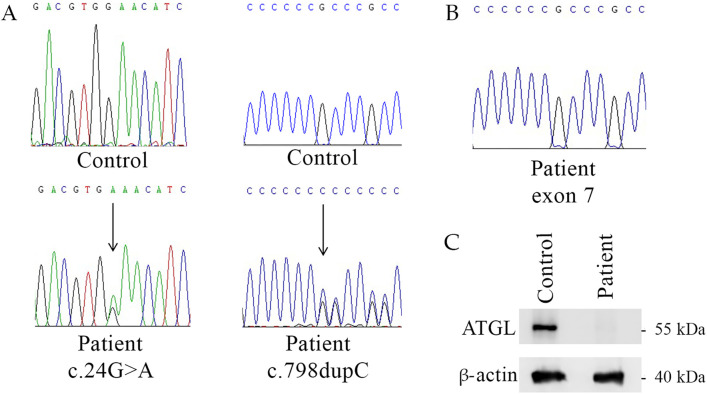
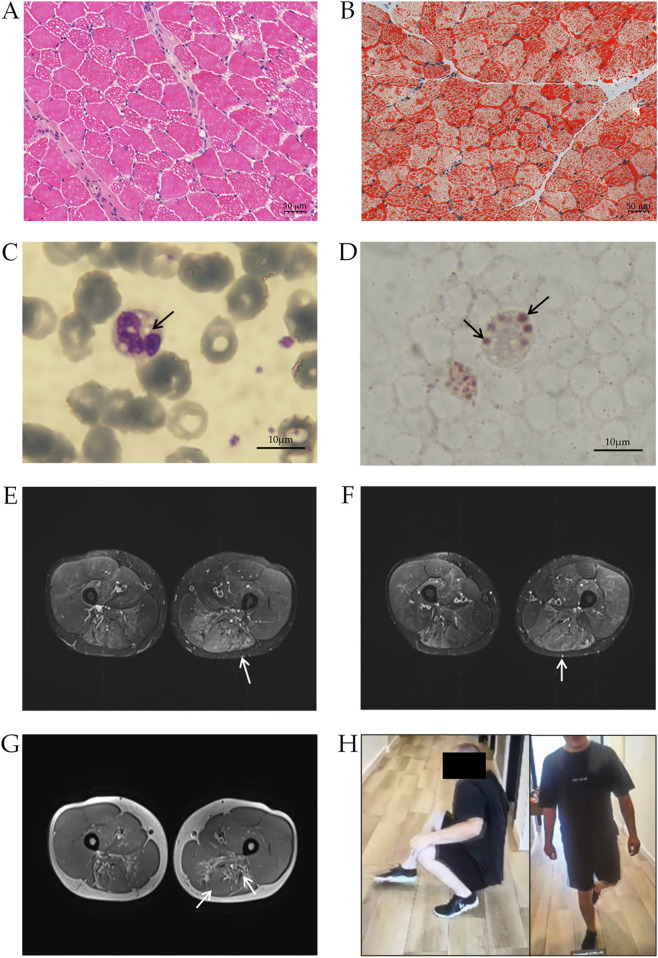
Neutral Lipid Storage Disease with Myopathy (NLSDM) is a rare lipid metabolism disorder caused by impaired Adipose Triglyceride Lipase (ATGL) activity, leading to neutral lipid accumulation in various tissues. It typically manifests with progressive skeletal myopathy, with an onset of around 35 years. In addition, some patients develop cardiomyopathy and liver dysfunction. Herein, we report the molecular characterization of a 27-year-old Hungarian patient and his family in whom two severe PNPLA2 mutations led to complete loss of ATGL production in the patient's tissues. DNA sequencing revealed a nonsense (c.24G>A) and a frameshift mutation (c.798dupC) in the PNPLA2 gene. RNA analysis showed nonsense-mediated decay of the c.798dupC transcript, while c.24G>A was normally expressed in the patient. However, Western blot analysis did not detect ATGL protein production. From a clinical perspective, our patient exhibited pes planus, proximal muscle weakness of the lower limbs and elevated CK levels from the age of six. MRI and biopsy revealed lipid accumulation, and leukocytes showed Jordans' anomaly. The muscle weakness progressed, and cardiomyopathy and hepatic steatosis were also observed recently. The case highlights two severe PNPLA2 mutations leading to complete ATGL deficiency and an unusual early-onset myopathy in childhood.
Frontiers in GeneticsBiochemistry, Genetics and Molecular Biology-Molecular Medicine
CiteScore
5.50
自引率
8.10%
发文量
3491
审稿时长
14 weeks
期刊介绍:
Frontiers in Genetics publishes rigorously peer-reviewed research on genes and genomes relating to all the domains of life, from humans to plants to livestock and other model organisms. Led by an outstanding Editorial Board of the world’s leading experts, this multidisciplinary, open-access journal is at the forefront of communicating cutting-edge research to researchers, academics, clinicians, policy makers and the public.
The study of inheritance and the impact of the genome on various biological processes is well documented. However, the majority of discoveries are still to come. A new era is seeing major developments in the function and variability of the genome, the use of genetic and genomic tools and the analysis of the genetic basis of various biological phenomena.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


