{"title":"基于蛋白质语言嵌入和物理化学信息的注意图引导图卷积网络预测核酸结合位点。","authors":"Xiang Li, Wei Peng, Xiaolei Zhu","doi":"10.1093/bib/bbaf457","DOIUrl":null,"url":null,"abstract":"<p><p>Protein-nucleic acid binding sites play a crucial role in biological processes such as gene expression, signal transduction, replication, and transcription. In recent years, with the development of artificial intelligence, protein language models, graph neural networks, and transformer architectures have been adopted to develop both structure-based and sequence-based predictive models. Structure-based methods benefit from the spatial relationship between residues and have shown promising performance. However, structure-based information requires 3D protein structures, which is a challenge for large-scale protein sequence spaces. To address this limitation, researchers have attempted to use predicted protein structure information to guide binding site prediction. While this strategy has improved accuracy, it still depends on the quality of structure predictions. Thus, some studies have returned to prediction methods based solely on protein sequences, particularly those using protein language models, which have greatly enhanced the prediction accuracy. This paper proposes a novel protein-nucleic acid binding site prediction framework, ATtention Maps and Graph convolutional neural networks to predict nucleic acid-protein Binding sites (ATMGBs), which first fuses protein language embeddings with physicochemical properties to obtain multiview information, then leverages the attention map of a protein language model to simulate the relationship between residues, and then utilizes graph convolutional networks for enhancing the feature representations for final prediction. ATMGBs was evaluated on several different independent test sets. The results indicate that the proposed approach significantly improves sequence-based prediction performance, even achieving prediction accuracy comparable to structure-based frameworks. The dataset and code used in this study are available at https://github.com/lixiangli01/ATMGBs.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"26 5","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12415854/pdf/","citationCount":"0","resultStr":"{\"title\":\"Predicting nucleic acid binding sites by attention map-guided graph convolutional network with protein language embeddings and physicochemical information.\",\"authors\":\"Xiang Li, Wei Peng, Xiaolei Zhu\",\"doi\":\"10.1093/bib/bbaf457\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Protein-nucleic acid binding sites play a crucial role in biological processes such as gene expression, signal transduction, replication, and transcription. In recent years, with the development of artificial intelligence, protein language models, graph neural networks, and transformer architectures have been adopted to develop both structure-based and sequence-based predictive models. Structure-based methods benefit from the spatial relationship between residues and have shown promising performance. However, structure-based information requires 3D protein structures, which is a challenge for large-scale protein sequence spaces. To address this limitation, researchers have attempted to use predicted protein structure information to guide binding site prediction. While this strategy has improved accuracy, it still depends on the quality of structure predictions. Thus, some studies have returned to prediction methods based solely on protein sequences, particularly those using protein language models, which have greatly enhanced the prediction accuracy. This paper proposes a novel protein-nucleic acid binding site prediction framework, ATtention Maps and Graph convolutional neural networks to predict nucleic acid-protein Binding sites (ATMGBs), which first fuses protein language embeddings with physicochemical properties to obtain multiview information, then leverages the attention map of a protein language model to simulate the relationship between residues, and then utilizes graph convolutional networks for enhancing the feature representations for final prediction. ATMGBs was evaluated on several different independent test sets. The results indicate that the proposed approach significantly improves sequence-based prediction performance, even achieving prediction accuracy comparable to structure-based frameworks. The dataset and code used in this study are available at https://github.com/lixiangli01/ATMGBs.</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"26 5\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2025-08-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12415854/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbaf457\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbaf457","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Predicting nucleic acid binding sites by attention map-guided graph convolutional network with protein language embeddings and physicochemical information.
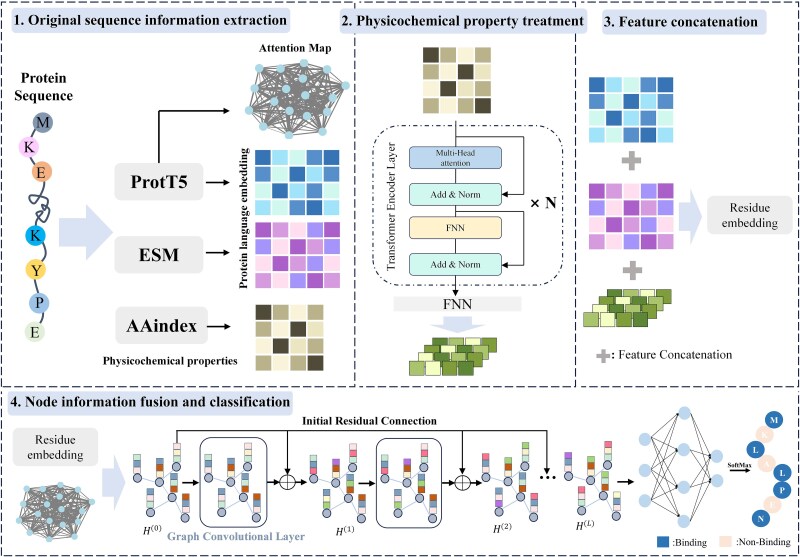
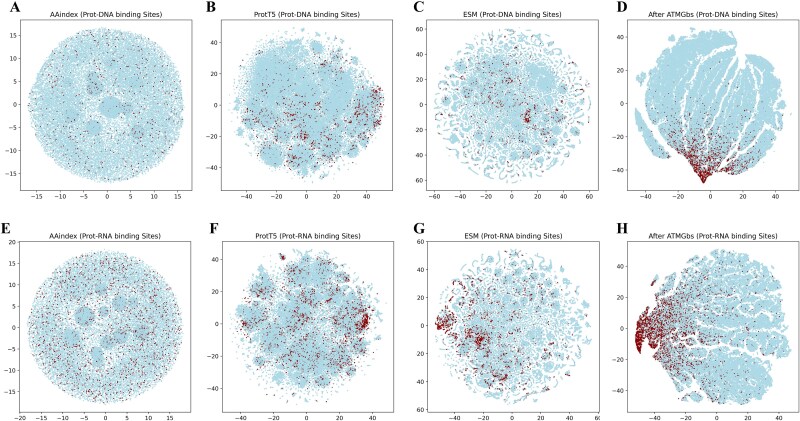
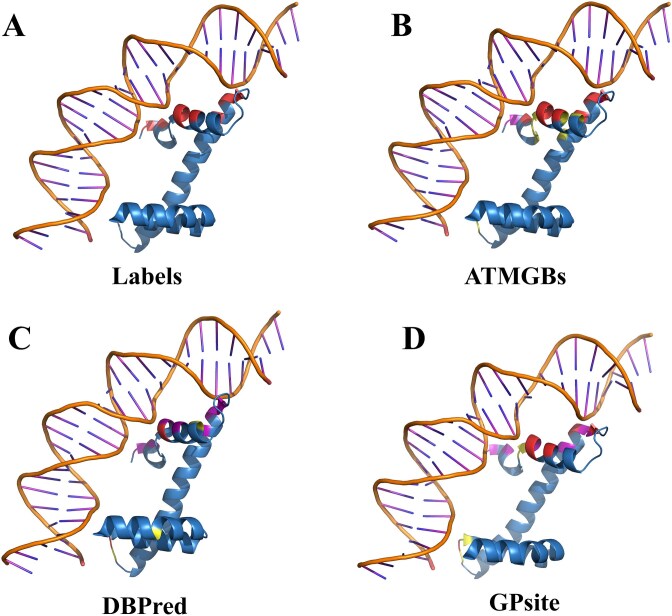
Protein-nucleic acid binding sites play a crucial role in biological processes such as gene expression, signal transduction, replication, and transcription. In recent years, with the development of artificial intelligence, protein language models, graph neural networks, and transformer architectures have been adopted to develop both structure-based and sequence-based predictive models. Structure-based methods benefit from the spatial relationship between residues and have shown promising performance. However, structure-based information requires 3D protein structures, which is a challenge for large-scale protein sequence spaces. To address this limitation, researchers have attempted to use predicted protein structure information to guide binding site prediction. While this strategy has improved accuracy, it still depends on the quality of structure predictions. Thus, some studies have returned to prediction methods based solely on protein sequences, particularly those using protein language models, which have greatly enhanced the prediction accuracy. This paper proposes a novel protein-nucleic acid binding site prediction framework, ATtention Maps and Graph convolutional neural networks to predict nucleic acid-protein Binding sites (ATMGBs), which first fuses protein language embeddings with physicochemical properties to obtain multiview information, then leverages the attention map of a protein language model to simulate the relationship between residues, and then utilizes graph convolutional networks for enhancing the feature representations for final prediction. ATMGBs was evaluated on several different independent test sets. The results indicate that the proposed approach significantly improves sequence-based prediction performance, even achieving prediction accuracy comparable to structure-based frameworks. The dataset and code used in this study are available at https://github.com/lixiangli01/ATMGBs.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


