评估用于准确预测冠心病核小体重塑者致病性的计算机工具。
IF 4.5
2区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
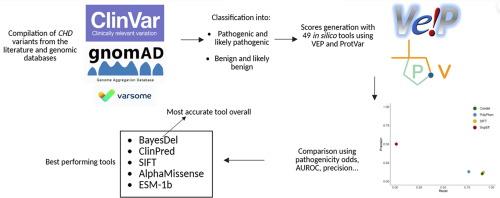
染色质结构域解旋酶DNA结合(CHD)蛋白是染色质重塑蛋白家族的组成部分,在DNA修复和基因表达调控、神经干细胞分化和染色质完整性中起着至关重要的作用。冠心病染色质重塑基因的遗传变异与自闭症谱系障碍和智力残疾等神经发育障碍有关。因此,在临床基因检测中确定冠心病变异个体的变异致病性是至关重要的。在这项研究中,我们比较了多种致病性预测工具的效率,这些工具是识别和注释潜在致病变异的宝贵资源,以评估能够区分致病性冠心病变异和良性冠心病变异的最准确的计算机工具。我们特别关注那些具有高度结构和功能相似性并与致病性突变密切相关的基因。在这里,我们评估了一系列致病性预测工具,并将其输出与文献和基因组数据库中报道的致病性结论进行了比较。我们的研究结果显示,表现最好的工具是BayesDel, ClinPred, AlphaMissense, ESM-1b和SIFT。BayesDel,特别是其addAF成分,总体上是预测冠心病变异致病性最可靠的工具。我们还建议将SnpEff的高影响变异识别能力用于开发混合工具,以增强冠心病变异的分类。我们的研究强调需要持续评估和整合更新的预测工具,包括新兴的人工智能方法。本研究还强调了收集更好的临床和机制数据对致病变异的危害,以提高临床诊断的准确性的重要性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Assessing In Silico Tools for Accurate Pathogenicity Prediction in CHD Nucleosome Remodelers
Chromodomain Helicase DNA-binding (CHD) proteins compose a family of chromatin remodelers that play crucial roles in DNA repair, gene expression regulation, neural stem cell differentiation and chromatin integrity. Genetic variants in CHD chromatin remodelers are associated with neurodevelopmental disorders with features like autism spectrum disorder and intellectual disability. Consequently, the determination of variant pathogenicity in clinical genetic tests for individuals bearing CHD variants is crucial. In this study, we compared the efficiency of multiple pathogenicity prediction tools, which are valuable resources for the identification and annotation of potentially disease-causing variants, to assess the most accurate in silico tool capable of distinguishing pathogenic CHD variants from benign ones. We have focused specifically on genes that share high structural and functional similarity and are strongly linked to pathogenic mutations. Here, we evaluated a range of pathogenicity prediction tools and compared their output with pathogenicity conclusions reported in the literature and genomic databases. Our findings showed that the top performing tools were BayesDel, ClinPred, AlphaMissense, ESM-1b and SIFT. BayesDel, specifically with its addAF component, was overall the most robust tool for CHD variant pathogenicity prediction. We also suggest incorporating SnpEff’s high-impact variant identification capabilities for the development of a hybrid tool that would enhance the classification of CHD variants. Our study emphasizes the need for continuous evaluation and integration of updated prediction tools, including emerging artificial intelligence (AI) approaches. This research also emphasizes the importance of gathering better clinical and mechanistic data on the deleteriousness of pathogenic variants to improve the accuracy of clinical diagnostics.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Molecular Biology
生物-生化与分子生物学
CiteScore
11.30
自引率
1.80%
发文量
412
审稿时长
28 days
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


