Qicheng Bei, Nathan L R Williams, Laura E Furtado, Daria Di Blasi, Jelani Williams, Vanda Brotas, Glen Tarran, Andrew P Rees, Chris Bowler, Jed A Fuhrman
{"title":"海洋原核生物和光合真核生物的定量宏基因组学。","authors":"Qicheng Bei, Nathan L R Williams, Laura E Furtado, Daria Di Blasi, Jelani Williams, Vanda Brotas, Glen Tarran, Andrew P Rees, Chris Bowler, Jed A Fuhrman","doi":"10.1093/ismeco/ycaf131","DOIUrl":null,"url":null,"abstract":"<p><p>High-throughput sequencing has provided unprecedented insights into microbial biodiversity in marine and other ecosystems. However, most sequencing-based studies report only relative (compositional) rather than absolute abundance, limiting their application in ecological modeling and biogeochemical analyses. Here, we present a metagenomic protocol incorporating genomic internal standards to quantify the absolute abundances of prokaryotes and eukaryotic phytoplankton, which together form the base of the marine food web, in unfractionated seawater. We applied this method to surface waters collected across 50°N to 40°S during the 29<sup>th</sup> Atlantic Meridional Transect. Using the single-copy <i>recA</i> gene, we estimated an average bacterial abundance of 1.0 × 10<sup>9</sup> haploid genome equivalents per liter. Leveraging a recent report that the <i>psbO</i> gene is typically single-copy in phytoplankton, we also quantified eukaryotic phytoplankton. Metagenomic estimates closely aligned with flow cytometry data for cyanobacteria (slope = 1.03, Pearson's <i>r</i> = 0.89) and eukaryotic phytoplankton (slope = 0.72, Pearson's <i>r</i> = 0.84). Compared to flow cytometry, taxonomic resolution for nano- and picoeukaryotes was greatly improved. Estimates for diatoms, dinoflagellates, and <i>Trichodesmium</i> were considerably higher than microscopy counts, likely reflecting microscopy undercounts and potential ploidy variation. These findings highlight the value of absolute quantification by metagenomics and offer a robust framework for quantitative assessments in microbial oceanography.</p>","PeriodicalId":73516,"journal":{"name":"ISME communications","volume":"5 1","pages":"ycaf131"},"PeriodicalIF":6.1000,"publicationDate":"2025-07-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12378644/pdf/","citationCount":"0","resultStr":"{\"title\":\"Quantitative metagenomics for marine prokaryotes and photosynthetic eukaryotes.\",\"authors\":\"Qicheng Bei, Nathan L R Williams, Laura E Furtado, Daria Di Blasi, Jelani Williams, Vanda Brotas, Glen Tarran, Andrew P Rees, Chris Bowler, Jed A Fuhrman\",\"doi\":\"10.1093/ismeco/ycaf131\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>High-throughput sequencing has provided unprecedented insights into microbial biodiversity in marine and other ecosystems. However, most sequencing-based studies report only relative (compositional) rather than absolute abundance, limiting their application in ecological modeling and biogeochemical analyses. Here, we present a metagenomic protocol incorporating genomic internal standards to quantify the absolute abundances of prokaryotes and eukaryotic phytoplankton, which together form the base of the marine food web, in unfractionated seawater. We applied this method to surface waters collected across 50°N to 40°S during the 29<sup>th</sup> Atlantic Meridional Transect. Using the single-copy <i>recA</i> gene, we estimated an average bacterial abundance of 1.0 × 10<sup>9</sup> haploid genome equivalents per liter. Leveraging a recent report that the <i>psbO</i> gene is typically single-copy in phytoplankton, we also quantified eukaryotic phytoplankton. Metagenomic estimates closely aligned with flow cytometry data for cyanobacteria (slope = 1.03, Pearson's <i>r</i> = 0.89) and eukaryotic phytoplankton (slope = 0.72, Pearson's <i>r</i> = 0.84). Compared to flow cytometry, taxonomic resolution for nano- and picoeukaryotes was greatly improved. Estimates for diatoms, dinoflagellates, and <i>Trichodesmium</i> were considerably higher than microscopy counts, likely reflecting microscopy undercounts and potential ploidy variation. These findings highlight the value of absolute quantification by metagenomics and offer a robust framework for quantitative assessments in microbial oceanography.</p>\",\"PeriodicalId\":73516,\"journal\":{\"name\":\"ISME communications\",\"volume\":\"5 1\",\"pages\":\"ycaf131\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2025-07-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12378644/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISME communications\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/ismeco/ycaf131\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"ECOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISME communications","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/ismeco/ycaf131","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"ECOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
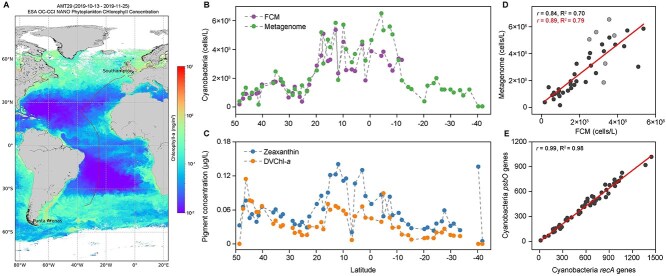
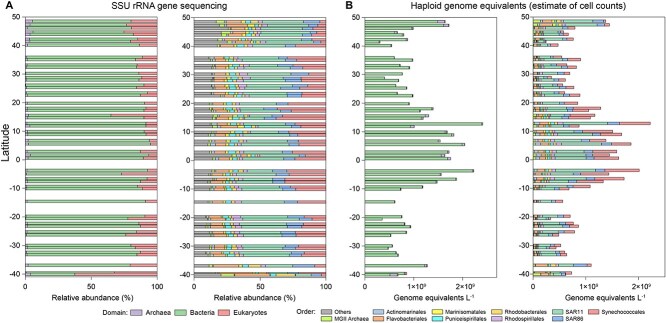
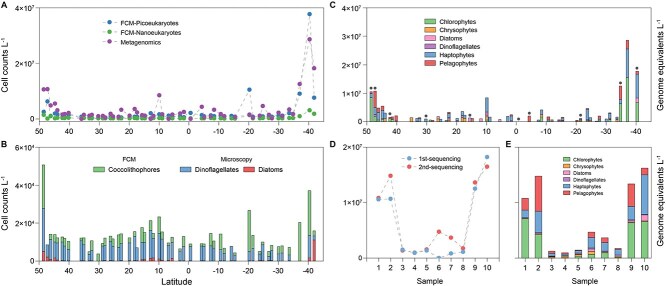
高通量测序为海洋和其他生态系统中的微生物生物多样性提供了前所未有的见解。然而,大多数基于测序的研究只报告了相对(成分)丰度,而不是绝对丰度,限制了它们在生态建模和生物地球化学分析中的应用。在这里,我们提出了一个包含基因组内部标准的宏基因组方案,以量化原核生物和真核浮游植物的绝对丰度,它们共同构成了海洋食物网的基础,在未分离的海水中。我们将这种方法应用于第29大西洋经向样带期间从50°N到40°S收集的地表水。使用单拷贝recA基因,我们估计平均细菌丰度为每升1.0 × 109个单倍体基因组当量。利用最近的一份报告,psbO基因在浮游植物中通常是单拷贝的,我们也量化了真核浮游植物。对于蓝藻(斜率= 1.03,Pearson’s r = 0.89)和真核浮游植物(斜率= 0.72,Pearson’s r = 0.84)的元基因组估计与流式细胞术数据非常接近。与流式细胞术相比,纳米和微真核细胞的分类分辨率大大提高。硅藻、鞭毛藻和毛菌的估计值明显高于显微镜计数,可能反映了显微镜计数不足和潜在的倍性变异。这些发现突出了宏基因组学绝对定量的价值,并为微生物海洋学的定量评估提供了一个强有力的框架。
Quantitative metagenomics for marine prokaryotes and photosynthetic eukaryotes.
High-throughput sequencing has provided unprecedented insights into microbial biodiversity in marine and other ecosystems. However, most sequencing-based studies report only relative (compositional) rather than absolute abundance, limiting their application in ecological modeling and biogeochemical analyses. Here, we present a metagenomic protocol incorporating genomic internal standards to quantify the absolute abundances of prokaryotes and eukaryotic phytoplankton, which together form the base of the marine food web, in unfractionated seawater. We applied this method to surface waters collected across 50°N to 40°S during the 29th Atlantic Meridional Transect. Using the single-copy recA gene, we estimated an average bacterial abundance of 1.0 × 109 haploid genome equivalents per liter. Leveraging a recent report that the psbO gene is typically single-copy in phytoplankton, we also quantified eukaryotic phytoplankton. Metagenomic estimates closely aligned with flow cytometry data for cyanobacteria (slope = 1.03, Pearson's r = 0.89) and eukaryotic phytoplankton (slope = 0.72, Pearson's r = 0.84). Compared to flow cytometry, taxonomic resolution for nano- and picoeukaryotes was greatly improved. Estimates for diatoms, dinoflagellates, and Trichodesmium were considerably higher than microscopy counts, likely reflecting microscopy undercounts and potential ploidy variation. These findings highlight the value of absolute quantification by metagenomics and offer a robust framework for quantitative assessments in microbial oceanography.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


