Adelaide Ambrosio, Tiziana Fioretti, Barbara D'Andrea, Lucia Pezone, Ilaria Bitetti, Carmela Di Domenico, Sabrina Vallone, Valeria Maiolo, Angela Cioce, Mariano Giustino, Antonio Varone, Gabriella Esposito
{"title":"坎帕尼亚地区(意大利)新生儿脊髓性肌萎缩症筛查项目:目前的局限性和潜在的前景。","authors":"Adelaide Ambrosio, Tiziana Fioretti, Barbara D'Andrea, Lucia Pezone, Ilaria Bitetti, Carmela Di Domenico, Sabrina Vallone, Valeria Maiolo, Angela Cioce, Mariano Giustino, Antonio Varone, Gabriella Esposito","doi":"10.3390/ijns11030064","DOIUrl":null,"url":null,"abstract":"<p><p>Three targeted therapies are currently available for spinal muscular atrophy (SMA), which have dramatically changed the natural history of this severe and potentially fatal disease. More than 95% of SMA cases have a homozygous deletion of exon 7 of the <i>SMN1</i> gene. Disease expression mainly depends on the copy number of <i>SMN2</i>, a hypomorphic copy of <i>SMN1</i>. Many countries in the world have implemented newborn screening (NBS) programs for early identification and treatment of children with SMA. We herein present the first two-year results of the SMA NBS program in Campania, a region with one of the highest birth rates in Italy. Genomic DNA was extracted from dried blood spots (DBS) and peripheral blood. For DBS, the <i>SMN1</i> gene copy number was evaluated by quantitative polymerase chain reaction (qPCR) targeting <i>SMN1</i> exon 7 and a reference gene (<i>RPP30</i>). In positive newborns and their parents, <i>SMN1</i>/<i>SMN2</i> copies were evaluated by multiplex ligation probe amplification (MLPA). We analyzed 77,945 newborns and identified 11 positive children. Six patients had 2 copies of <i>SMN2</i>, but only one showed severe SMA-related signs at birth. Eligible newborns were treated with gene therapy within 20 days of birth. Notably, qPCR failed to amplify the reference <i>RPP30</i> gene in 10/77,945 DBS. Despite this limitation, we observed that about 1/40 DBS had ΔCt values consistent with the presence of one <i>SMN1</i> copy. The semi-automated procedure used for SMA NBS showed excellent performance in detecting the presence of homozygous deletion of <i>SMN1</i> exon 7, with the exception of a few cases with the absence of amplification of the reference gene. By solving this limitation, the screening procedure has the potential to detect heterozygous carriers of the <i>SMN1</i> deletion and, consequently, identify families at procreative risk of SMA.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 3","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12371891/pdf/","citationCount":"0","resultStr":"{\"title\":\"Newborn Screening Program for Spinal Muscular Atrophy in the Campania Region (Italy): Current Limitations and Potential Perspectives.\",\"authors\":\"Adelaide Ambrosio, Tiziana Fioretti, Barbara D'Andrea, Lucia Pezone, Ilaria Bitetti, Carmela Di Domenico, Sabrina Vallone, Valeria Maiolo, Angela Cioce, Mariano Giustino, Antonio Varone, Gabriella Esposito\",\"doi\":\"10.3390/ijns11030064\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Three targeted therapies are currently available for spinal muscular atrophy (SMA), which have dramatically changed the natural history of this severe and potentially fatal disease. More than 95% of SMA cases have a homozygous deletion of exon 7 of the <i>SMN1</i> gene. Disease expression mainly depends on the copy number of <i>SMN2</i>, a hypomorphic copy of <i>SMN1</i>. Many countries in the world have implemented newborn screening (NBS) programs for early identification and treatment of children with SMA. We herein present the first two-year results of the SMA NBS program in Campania, a region with one of the highest birth rates in Italy. Genomic DNA was extracted from dried blood spots (DBS) and peripheral blood. For DBS, the <i>SMN1</i> gene copy number was evaluated by quantitative polymerase chain reaction (qPCR) targeting <i>SMN1</i> exon 7 and a reference gene (<i>RPP30</i>). In positive newborns and their parents, <i>SMN1</i>/<i>SMN2</i> copies were evaluated by multiplex ligation probe amplification (MLPA). We analyzed 77,945 newborns and identified 11 positive children. Six patients had 2 copies of <i>SMN2</i>, but only one showed severe SMA-related signs at birth. Eligible newborns were treated with gene therapy within 20 days of birth. Notably, qPCR failed to amplify the reference <i>RPP30</i> gene in 10/77,945 DBS. Despite this limitation, we observed that about 1/40 DBS had ΔCt values consistent with the presence of one <i>SMN1</i> copy. The semi-automated procedure used for SMA NBS showed excellent performance in detecting the presence of homozygous deletion of <i>SMN1</i> exon 7, with the exception of a few cases with the absence of amplification of the reference gene. By solving this limitation, the screening procedure has the potential to detect heterozygous carriers of the <i>SMN1</i> deletion and, consequently, identify families at procreative risk of SMA.</p>\",\"PeriodicalId\":14159,\"journal\":{\"name\":\"International Journal of Neonatal Screening\",\"volume\":\"11 3\",\"pages\":\"\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2025-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12371891/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Neonatal Screening\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/ijns11030064\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11030064","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Newborn Screening Program for Spinal Muscular Atrophy in the Campania Region (Italy): Current Limitations and Potential Perspectives.
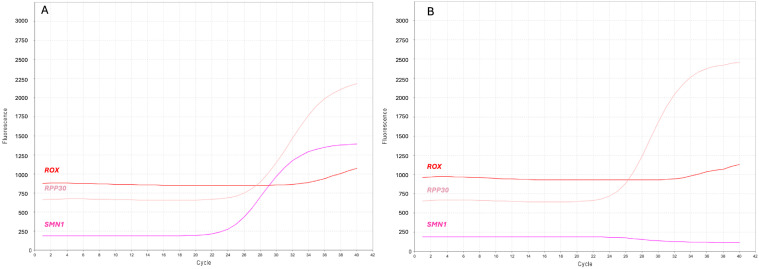
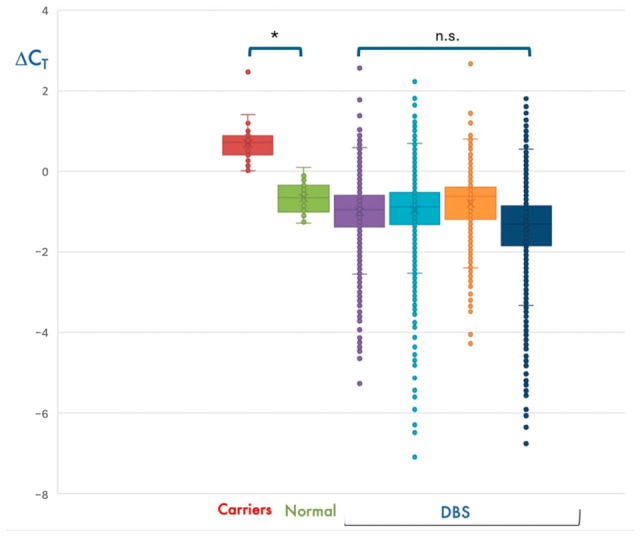
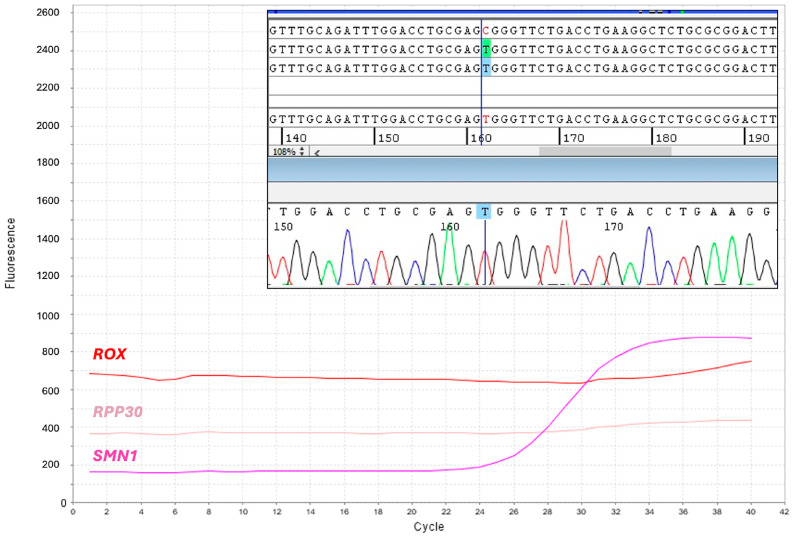
Three targeted therapies are currently available for spinal muscular atrophy (SMA), which have dramatically changed the natural history of this severe and potentially fatal disease. More than 95% of SMA cases have a homozygous deletion of exon 7 of the SMN1 gene. Disease expression mainly depends on the copy number of SMN2, a hypomorphic copy of SMN1. Many countries in the world have implemented newborn screening (NBS) programs for early identification and treatment of children with SMA. We herein present the first two-year results of the SMA NBS program in Campania, a region with one of the highest birth rates in Italy. Genomic DNA was extracted from dried blood spots (DBS) and peripheral blood. For DBS, the SMN1 gene copy number was evaluated by quantitative polymerase chain reaction (qPCR) targeting SMN1 exon 7 and a reference gene (RPP30). In positive newborns and their parents, SMN1/SMN2 copies were evaluated by multiplex ligation probe amplification (MLPA). We analyzed 77,945 newborns and identified 11 positive children. Six patients had 2 copies of SMN2, but only one showed severe SMA-related signs at birth. Eligible newborns were treated with gene therapy within 20 days of birth. Notably, qPCR failed to amplify the reference RPP30 gene in 10/77,945 DBS. Despite this limitation, we observed that about 1/40 DBS had ΔCt values consistent with the presence of one SMN1 copy. The semi-automated procedure used for SMA NBS showed excellent performance in detecting the presence of homozygous deletion of SMN1 exon 7, with the exception of a few cases with the absence of amplification of the reference gene. By solving this limitation, the screening procedure has the potential to detect heterozygous carriers of the SMN1 deletion and, consequently, identify families at procreative risk of SMA.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


