Hans-Christof Gasser, Ajitha Rajan, Javier A Alfaro
{"title":"一种新的解码策略,用于设计对细胞毒性t淋巴细胞可视性较低的ProteinMPNN。","authors":"Hans-Christof Gasser, Ajitha Rajan, Javier A Alfaro","doi":"10.1016/j.csbj.2025.07.055","DOIUrl":null,"url":null,"abstract":"<p><p>Due to their versatility and diverse production methods, proteins have attracted a lot of interest for industrial as well as therapeutic applications. Designing new therapeutics requires careful consideration of immune responses, particularly the cytotoxic T-lymphocyte (CTL) reaction to intra-cellular proteins. In this study, we introduce CAPE-Beam, a novel decoding strategy for the established ProteinMPNN protein design model. Our approach minimizes CTL immunogenicity risk by limiting designs to only consist of kmers that are either predicted not to be presented to CTLs or are subject to central tolerance that prevents CTLs from attacking self-peptides. We compare CAPE-Beam to the standard way of sampling from ProteinMPNN and the state of the art (SOTA) technique CAPE-MPNN. We find that our novel decoding strategy can produce structurally similar proteins while incorporating more human like kmers. This significantly lowers CTL immunogenicity risk in precision medicine, and represents a key step towards reducing this risk in protein therapeutics targeting a wider patient population. Source: https://github.com/hcgasser/CAPE_Beam.</p>","PeriodicalId":10715,"journal":{"name":"Computational and structural biotechnology journal","volume":"27 ","pages":"3693-3703"},"PeriodicalIF":4.1000,"publicationDate":"2025-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12396444/pdf/","citationCount":"0","resultStr":"{\"title\":\"A novel decoding strategy for ProteinMPNN to design with less visibility to cytotoxic T-lymphocytes.\",\"authors\":\"Hans-Christof Gasser, Ajitha Rajan, Javier A Alfaro\",\"doi\":\"10.1016/j.csbj.2025.07.055\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Due to their versatility and diverse production methods, proteins have attracted a lot of interest for industrial as well as therapeutic applications. Designing new therapeutics requires careful consideration of immune responses, particularly the cytotoxic T-lymphocyte (CTL) reaction to intra-cellular proteins. In this study, we introduce CAPE-Beam, a novel decoding strategy for the established ProteinMPNN protein design model. Our approach minimizes CTL immunogenicity risk by limiting designs to only consist of kmers that are either predicted not to be presented to CTLs or are subject to central tolerance that prevents CTLs from attacking self-peptides. We compare CAPE-Beam to the standard way of sampling from ProteinMPNN and the state of the art (SOTA) technique CAPE-MPNN. We find that our novel decoding strategy can produce structurally similar proteins while incorporating more human like kmers. This significantly lowers CTL immunogenicity risk in precision medicine, and represents a key step towards reducing this risk in protein therapeutics targeting a wider patient population. Source: https://github.com/hcgasser/CAPE_Beam.</p>\",\"PeriodicalId\":10715,\"journal\":{\"name\":\"Computational and structural biotechnology journal\",\"volume\":\"27 \",\"pages\":\"3693-3703\"},\"PeriodicalIF\":4.1000,\"publicationDate\":\"2025-08-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12396444/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and structural biotechnology journal\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.csbj.2025.07.055\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and structural biotechnology journal","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.csbj.2025.07.055","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
A novel decoding strategy for ProteinMPNN to design with less visibility to cytotoxic T-lymphocytes.
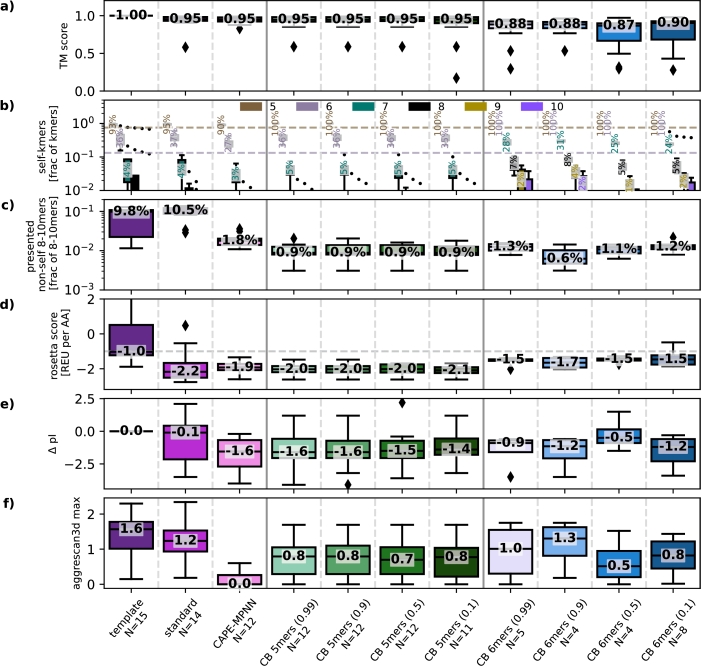
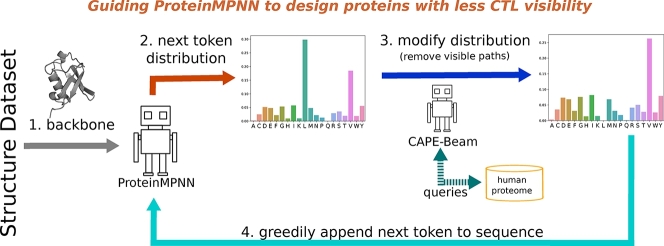
Due to their versatility and diverse production methods, proteins have attracted a lot of interest for industrial as well as therapeutic applications. Designing new therapeutics requires careful consideration of immune responses, particularly the cytotoxic T-lymphocyte (CTL) reaction to intra-cellular proteins. In this study, we introduce CAPE-Beam, a novel decoding strategy for the established ProteinMPNN protein design model. Our approach minimizes CTL immunogenicity risk by limiting designs to only consist of kmers that are either predicted not to be presented to CTLs or are subject to central tolerance that prevents CTLs from attacking self-peptides. We compare CAPE-Beam to the standard way of sampling from ProteinMPNN and the state of the art (SOTA) technique CAPE-MPNN. We find that our novel decoding strategy can produce structurally similar proteins while incorporating more human like kmers. This significantly lowers CTL immunogenicity risk in precision medicine, and represents a key step towards reducing this risk in protein therapeutics targeting a wider patient population. Source: https://github.com/hcgasser/CAPE_Beam.
期刊介绍:
Computational and Structural Biotechnology Journal (CSBJ) is an online gold open access journal publishing research articles and reviews after full peer review. All articles are published, without barriers to access, immediately upon acceptance. The journal places a strong emphasis on functional and mechanistic understanding of how molecular components in a biological process work together through the application of computational methods. Structural data may provide such insights, but they are not a pre-requisite for publication in the journal. Specific areas of interest include, but are not limited to:
Structure and function of proteins, nucleic acids and other macromolecules
Structure and function of multi-component complexes
Protein folding, processing and degradation
Enzymology
Computational and structural studies of plant systems
Microbial Informatics
Genomics
Proteomics
Metabolomics
Algorithms and Hypothesis in Bioinformatics
Mathematical and Theoretical Biology
Computational Chemistry and Drug Discovery
Microscopy and Molecular Imaging
Nanotechnology
Systems and Synthetic Biology

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


