Lisa Lombardo, , , Verena Battisti, , , Thierry Langer, , , Rosaria Gitto, , and , Laura De Luca*,
{"title":"利用机器学习方法预测Sigma受体配体的活性和选择性","authors":"Lisa Lombardo, , , Verena Battisti, , , Thierry Langer, , , Rosaria Gitto, , and , Laura De Luca*, ","doi":"10.1021/acs.jcim.5c01091","DOIUrl":null,"url":null,"abstract":"<p >Sigma (σ) receptors (SRs) have emerged as important therapeutic targets due to their roles in various biological pathways. They are classified into two subtypes: S1R, primarily distributed in the central nervous system and related to neuroprotection and neurodegenerative diseases, and S2R mainly expressed in cancer cells and associated with cell proliferation and apoptosis, as well as in neurons. Although S1R and S2R exhibit structural differences in receptor architecture and assembly, they share similar binding site features and ligand recognition mechanisms. This similarity underscores the importance of identifying selective ligands for therapeutic design, especially given the distinct physiological functions of these receptors. In this project, we developed three distinct machine learning (ML) approaches based on classification, regression, and multiclassification models to predict the activity and selectivity profiles of SR ligands. High-quality data sets were curated from public and in-house source; in turn, the data sets were systematically organized and processed for each workflow. Models were built using molecular descriptors and fingerprints, including Mordred, RDKit, ECFP4, ECFP6, and MACCS keys, and trained with various ML algorithms such as extra trees, random forest, support vector machine, <i>k</i>-nearest neighbors, and XGBoost. Rigorous nested and classical 5-fold cross-validation protocols were applied for model selection and validation. At the end, identification of the best workflow was performed by an external validation procedure. Among the workflows, the one-step multiclassification approach, based on extra trees combined with Mordred descriptors, showed the best predictive performance in external validation, offering a robust tool for the identification of selective S1R and S2R ligands.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 18","pages":"9697–9712"},"PeriodicalIF":5.3000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jcim.5c01091","citationCount":"0","resultStr":"{\"title\":\"Prediction of Activity and Selectivity Profiles of Sigma Receptor Ligands Using Machine Learning Approaches\",\"authors\":\"Lisa Lombardo, , , Verena Battisti, , , Thierry Langer, , , Rosaria Gitto, , and , Laura De Luca*, \",\"doi\":\"10.1021/acs.jcim.5c01091\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Sigma (σ) receptors (SRs) have emerged as important therapeutic targets due to their roles in various biological pathways. They are classified into two subtypes: S1R, primarily distributed in the central nervous system and related to neuroprotection and neurodegenerative diseases, and S2R mainly expressed in cancer cells and associated with cell proliferation and apoptosis, as well as in neurons. Although S1R and S2R exhibit structural differences in receptor architecture and assembly, they share similar binding site features and ligand recognition mechanisms. This similarity underscores the importance of identifying selective ligands for therapeutic design, especially given the distinct physiological functions of these receptors. In this project, we developed three distinct machine learning (ML) approaches based on classification, regression, and multiclassification models to predict the activity and selectivity profiles of SR ligands. High-quality data sets were curated from public and in-house source; in turn, the data sets were systematically organized and processed for each workflow. Models were built using molecular descriptors and fingerprints, including Mordred, RDKit, ECFP4, ECFP6, and MACCS keys, and trained with various ML algorithms such as extra trees, random forest, support vector machine, <i>k</i>-nearest neighbors, and XGBoost. Rigorous nested and classical 5-fold cross-validation protocols were applied for model selection and validation. At the end, identification of the best workflow was performed by an external validation procedure. Among the workflows, the one-step multiclassification approach, based on extra trees combined with Mordred descriptors, showed the best predictive performance in external validation, offering a robust tool for the identification of selective S1R and S2R ligands.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 18\",\"pages\":\"9697–9712\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/acs.jcim.5c01091\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c01091\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c01091","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Prediction of Activity and Selectivity Profiles of Sigma Receptor Ligands Using Machine Learning Approaches
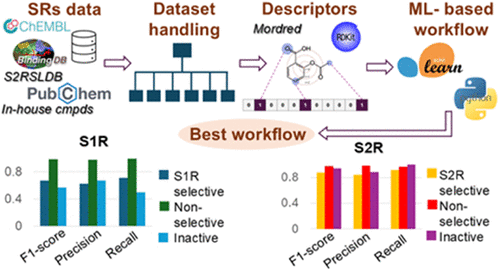
Sigma (σ) receptors (SRs) have emerged as important therapeutic targets due to their roles in various biological pathways. They are classified into two subtypes: S1R, primarily distributed in the central nervous system and related to neuroprotection and neurodegenerative diseases, and S2R mainly expressed in cancer cells and associated with cell proliferation and apoptosis, as well as in neurons. Although S1R and S2R exhibit structural differences in receptor architecture and assembly, they share similar binding site features and ligand recognition mechanisms. This similarity underscores the importance of identifying selective ligands for therapeutic design, especially given the distinct physiological functions of these receptors. In this project, we developed three distinct machine learning (ML) approaches based on classification, regression, and multiclassification models to predict the activity and selectivity profiles of SR ligands. High-quality data sets were curated from public and in-house source; in turn, the data sets were systematically organized and processed for each workflow. Models were built using molecular descriptors and fingerprints, including Mordred, RDKit, ECFP4, ECFP6, and MACCS keys, and trained with various ML algorithms such as extra trees, random forest, support vector machine, k-nearest neighbors, and XGBoost. Rigorous nested and classical 5-fold cross-validation protocols were applied for model selection and validation. At the end, identification of the best workflow was performed by an external validation procedure. Among the workflows, the one-step multiclassification approach, based on extra trees combined with Mordred descriptors, showed the best predictive performance in external validation, offering a robust tool for the identification of selective S1R and S2R ligands.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


