{"title":"Necdin的缺失通过SynGAP的不稳定导致社交缺陷和突触功能异常","authors":"Xiangyu Li, Ibrahim Bader, Xin Li, Renbin Lu, Dengfeng Liu, Zhiheng Chen, Suixin Deng, Yousheng Shu, Huadie Liu, Jing Zhang, Jia-Da Li","doi":"10.1038/s41380-025-03187-7","DOIUrl":null,"url":null,"abstract":"<p>The Ras GTPase-activating protein SynGAP interacts with PSD95 to regulate synaptic morphology and function at the postsynaptic density in neurons. Haploinsufficiency of <i>SYNGAP1</i> has been linked to autism spectrum disorders (ASD) and intellectual disability (ID). While transcriptional and translational regulation of SYNGAP1 has been extensively explored, the mechanisms governing its protein homeostasis remain largely elusive. In this study, we discovered that Necdin, a protein linked to Prader-Willi syndrome (PWS), interacts with SynGAP and regulates its stability through the SGT1-HSP90 chaperone machinery; notably, depletion of Necdin results in decreased SynGAP protein levels in mice. Loss of Necdin lead to impaired sociability, accompanied by an increased number of dendritic spines and a higher proportion of mature spines in pyramidal neurons of the medial prefrontal cortex (mPFC) in mice. Electrophysiological recordings revealed elevated frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) and reduced amplitude of miniature inhibitory postsynaptic currents (mIPSCs) in these neurons. Targeted viral overexpression of <i>Syngap1</i> in the mPFC of Necdin-deficient mice rescued the deficits in sociability, synaptic function, and dendritic spine morphology. Collectively, our findings reveal Necdin as a key regulator of SynGAP protein homeostasis and highlight the contribution of post-translational regulation in the pathogenesis of ASD.</p>","PeriodicalId":19008,"journal":{"name":"Molecular Psychiatry","volume":"26 1","pages":""},"PeriodicalIF":10.1000,"publicationDate":"2025-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Loss of Necdin causes social deficit and aberrant synaptic function through destabilization of SynGAP\",\"authors\":\"Xiangyu Li, Ibrahim Bader, Xin Li, Renbin Lu, Dengfeng Liu, Zhiheng Chen, Suixin Deng, Yousheng Shu, Huadie Liu, Jing Zhang, Jia-Da Li\",\"doi\":\"10.1038/s41380-025-03187-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The Ras GTPase-activating protein SynGAP interacts with PSD95 to regulate synaptic morphology and function at the postsynaptic density in neurons. Haploinsufficiency of <i>SYNGAP1</i> has been linked to autism spectrum disorders (ASD) and intellectual disability (ID). While transcriptional and translational regulation of SYNGAP1 has been extensively explored, the mechanisms governing its protein homeostasis remain largely elusive. In this study, we discovered that Necdin, a protein linked to Prader-Willi syndrome (PWS), interacts with SynGAP and regulates its stability through the SGT1-HSP90 chaperone machinery; notably, depletion of Necdin results in decreased SynGAP protein levels in mice. Loss of Necdin lead to impaired sociability, accompanied by an increased number of dendritic spines and a higher proportion of mature spines in pyramidal neurons of the medial prefrontal cortex (mPFC) in mice. Electrophysiological recordings revealed elevated frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) and reduced amplitude of miniature inhibitory postsynaptic currents (mIPSCs) in these neurons. Targeted viral overexpression of <i>Syngap1</i> in the mPFC of Necdin-deficient mice rescued the deficits in sociability, synaptic function, and dendritic spine morphology. Collectively, our findings reveal Necdin as a key regulator of SynGAP protein homeostasis and highlight the contribution of post-translational regulation in the pathogenesis of ASD.</p>\",\"PeriodicalId\":19008,\"journal\":{\"name\":\"Molecular Psychiatry\",\"volume\":\"26 1\",\"pages\":\"\"},\"PeriodicalIF\":10.1000,\"publicationDate\":\"2025-08-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Psychiatry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41380-025-03187-7\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Psychiatry","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41380-025-03187-7","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Loss of Necdin causes social deficit and aberrant synaptic function through destabilization of SynGAP
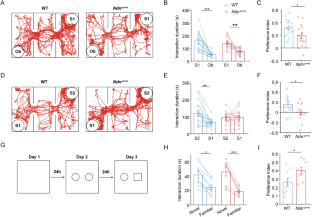
The Ras GTPase-activating protein SynGAP interacts with PSD95 to regulate synaptic morphology and function at the postsynaptic density in neurons. Haploinsufficiency of SYNGAP1 has been linked to autism spectrum disorders (ASD) and intellectual disability (ID). While transcriptional and translational regulation of SYNGAP1 has been extensively explored, the mechanisms governing its protein homeostasis remain largely elusive. In this study, we discovered that Necdin, a protein linked to Prader-Willi syndrome (PWS), interacts with SynGAP and regulates its stability through the SGT1-HSP90 chaperone machinery; notably, depletion of Necdin results in decreased SynGAP protein levels in mice. Loss of Necdin lead to impaired sociability, accompanied by an increased number of dendritic spines and a higher proportion of mature spines in pyramidal neurons of the medial prefrontal cortex (mPFC) in mice. Electrophysiological recordings revealed elevated frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) and reduced amplitude of miniature inhibitory postsynaptic currents (mIPSCs) in these neurons. Targeted viral overexpression of Syngap1 in the mPFC of Necdin-deficient mice rescued the deficits in sociability, synaptic function, and dendritic spine morphology. Collectively, our findings reveal Necdin as a key regulator of SynGAP protein homeostasis and highlight the contribution of post-translational regulation in the pathogenesis of ASD.
期刊介绍:
Molecular Psychiatry focuses on publishing research that aims to uncover the biological mechanisms behind psychiatric disorders and their treatment. The journal emphasizes studies that bridge pre-clinical and clinical research, covering cellular, molecular, integrative, clinical, imaging, and psychopharmacology levels.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


