利用机器学习和基于结构的药物设计原则相结合的方法来识别针对SphK1的潜在命中
IF 3.1
4区 生物学
Q2 BIOLOGY
引用次数: 0
摘要
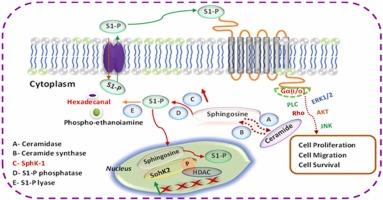
鞘氨醇激酶(SphK1)是一种至关重要的酶,它通过在鞘氨醇上添加一个磷酸基团,将其转化为鞘氨醇-1-磷酸,从而帮助鞘脂的加工。最近的一项研究表明,SphK1的失调与肺癌和膀胱癌的肿瘤进展和转移有关,使SphK1成为这些疾病的有希望的治疗靶点。在这项研究中,我们采用基于机器学习的虚拟筛选和基于结构的药物设计来识别具有不同化学支架的潜在SphK1抑制剂。利用分子指纹共生成16个机器学习模型,并利用最有效的模型对Maybridge图书馆进行虚拟筛选。然后对筛选的化合物进行分子对接,以确定针对SphK1蛋白的合适对接姿态。通过对最佳对接化合物的可视化分析,我们发现与对照(SQS)相比,有6种化合物与SphK1蛋白表现出强烈的相互作用。为了进一步支持我们的发现,我们对所有六种化合物进行了100 ns长的分子动力学(MD)模拟,以分析构象变化和稳定性。两种化合物(SCR00139和SCR00133)显示出有希望的稳定性,并且很适合SphK1蛋白的结合袋。此外,对这两种化合物进行了MM-PBSA和MM-GBSA研究,提供了良好的相对结合估计。本研究引入了一种基于机器学习的虚拟筛选的集成管道,用于识别针对癌症进展的新支架。然而,体外评价是必要的,以评估这些化合物的功效。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Utilizing a combined approach of machine learning and structure-based drug design principles to identify potential hits targeting SphK1
Sphingosine kinase (SphK1) is acrucial enzyme that aids in the processing of sphingolipids by adding a phosphate group to sphingosine, converting it into sphingosine-1-phosphate. A recent study has suggested that dysregulation of SphK1 is linked to tumor progression and metastasis in lung and bladder cancers,making SphK1 a promising therapeutic target for these diseases. In this study, we employedmachine learning-based virtual screening along with structure-based drug design to identify potential SphK1 inhibitors with diverse chemical scaffolds. A total of 16 machine learning models were generated using molecular fingerprints, and the most effective models were employed to conductvirtual screening of the Maybridge library. The screened compounds were then subjected to molecular docking to determine a suitable docked pose against the SphK1 protein. Upon visualization of the best docked compounds, we found that six compounds exhibited strong interactions with the SphK1 protein compared to the control (SQS). To further support our findings, we conducted 100 ns long molecular dynamics (MD) simulations of all six compounds to analyzeconformational changes and stability. Two compounds (SCR00139 and SCR00133) demonstratedpromising stability and fit well within the binding pocket of the SphK1 protein. Furthermore, MM-PBSA and MM-GBSA studies were carried out on these two compounds, providing favorable relative binding estimations. This study introduces an integrated pipeline of machine learning-based virtual screening for the identification of new scaffolds targeting cancer progression. However, in vitro evaluations are necessary to assess the efficacy of these compounds.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


