Peng Zhang, MengJie Bo, YuQin Chu, Yang Zhu, Peng Ma
{"title":"高氮杂环的静电场调制:能量和稳定性的计算研究","authors":"Peng Zhang, MengJie Bo, YuQin Chu, Yang Zhu, Peng Ma","doi":"10.1007/s00894-025-06482-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>In this study, the effect of external electric field (EEF) on three kinds of high nitrogen heterocyclic compounds with different ring structures (<b>PA-1 ~ 3</b>, oxadiazole, triazole, and tetrazole) was investigated by quantum chemical calculation. By using B3LYP/6-311G + (d, p) theory and Laplace bond order (LBO) analysis, the trigger bond was determined, and further EEF was applied along the <i>X</i>/<i>Y</i>/<i>Z</i> axis (0–0.02 a.u.) to study its effects on molecular structure, electron distribution, and DOS. The results show that EEF in different directions has a significant effect on charge transfer and intermolecular interaction, which provides a theoretical basis for regulating energy output and thermal stability. This work highlights the potential of computational strategies in the design of energetic materials with controllable properties, and builds a bridge between theoretical prediction and practical application in high-energy systems.</p><h3>Methods</h3><p>The Gaussian 16 software has been chosen for simulation and computation in this study. The B3LYP functional and 6-311G + (d, p) basis set has been utilized for the calculation and simulation of external electric fields, with the strength ranging from 0 to 0.020 a.u. and an increment gradient of 0.005 a.u.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 9","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Electrostatic field modulation of high-nitrogen heterocycles: a computational study on energy and stability\",\"authors\":\"Peng Zhang, MengJie Bo, YuQin Chu, Yang Zhu, Peng Ma\",\"doi\":\"10.1007/s00894-025-06482-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>In this study, the effect of external electric field (EEF) on three kinds of high nitrogen heterocyclic compounds with different ring structures (<b>PA-1 ~ 3</b>, oxadiazole, triazole, and tetrazole) was investigated by quantum chemical calculation. By using B3LYP/6-311G + (d, p) theory and Laplace bond order (LBO) analysis, the trigger bond was determined, and further EEF was applied along the <i>X</i>/<i>Y</i>/<i>Z</i> axis (0–0.02 a.u.) to study its effects on molecular structure, electron distribution, and DOS. The results show that EEF in different directions has a significant effect on charge transfer and intermolecular interaction, which provides a theoretical basis for regulating energy output and thermal stability. This work highlights the potential of computational strategies in the design of energetic materials with controllable properties, and builds a bridge between theoretical prediction and practical application in high-energy systems.</p><h3>Methods</h3><p>The Gaussian 16 software has been chosen for simulation and computation in this study. The B3LYP functional and 6-311G + (d, p) basis set has been utilized for the calculation and simulation of external electric fields, with the strength ranging from 0 to 0.020 a.u. and an increment gradient of 0.005 a.u.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"31 9\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-025-06482-w\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06482-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Electrostatic field modulation of high-nitrogen heterocycles: a computational study on energy and stability
Context
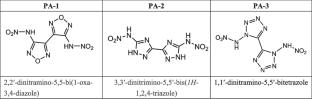
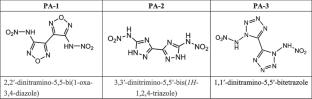
In this study, the effect of external electric field (EEF) on three kinds of high nitrogen heterocyclic compounds with different ring structures (PA-1 ~ 3, oxadiazole, triazole, and tetrazole) was investigated by quantum chemical calculation. By using B3LYP/6-311G + (d, p) theory and Laplace bond order (LBO) analysis, the trigger bond was determined, and further EEF was applied along the X/Y/Z axis (0–0.02 a.u.) to study its effects on molecular structure, electron distribution, and DOS. The results show that EEF in different directions has a significant effect on charge transfer and intermolecular interaction, which provides a theoretical basis for regulating energy output and thermal stability. This work highlights the potential of computational strategies in the design of energetic materials with controllable properties, and builds a bridge between theoretical prediction and practical application in high-energy systems.
Methods
The Gaussian 16 software has been chosen for simulation and computation in this study. The B3LYP functional and 6-311G + (d, p) basis set has been utilized for the calculation and simulation of external electric fields, with the strength ranging from 0 to 0.020 a.u. and an increment gradient of 0.005 a.u.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


