Karin Troell , Christen Rune Stensvold , Anna Rosa Sannella , Martha Betson , Emma Östlund , Rachel M. Chalmers , Umer Chaudhry , Rebecca Davidson , Lauren Davies , Ralf Ignatius , Anton de Jong , Gregory Karadjian , Karim Adjou , Christian Klotz , Sokratis Ptochos , Guy Robinson , Jeroen Roelfsema , Barbara Soba , Jacek Sroka , Paolo Vatta , Simone M. Cacciò
{"title":"一种新的人畜共患病原体小隐孢子虫多位点序列分型方案的设计、开发和测试","authors":"Karin Troell , Christen Rune Stensvold , Anna Rosa Sannella , Martha Betson , Emma Östlund , Rachel M. Chalmers , Umer Chaudhry , Rebecca Davidson , Lauren Davies , Ralf Ignatius , Anton de Jong , Gregory Karadjian , Karim Adjou , Christian Klotz , Sokratis Ptochos , Guy Robinson , Jeroen Roelfsema , Barbara Soba , Jacek Sroka , Paolo Vatta , Simone M. Cacciò","doi":"10.1016/j.crpvbd.2025.100308","DOIUrl":null,"url":null,"abstract":"<div><div>The zoonotic parasite <em>Cryptosporidium parvum</em> is an important global cause of diarrheal disease in humans and young ruminants. Molecular typing is essential to track transmission routes and identify clusters of cases. Here, we developed a novel multi-locus sequence typing (MLST) scheme based on single nucleotide polymorphisms (SNPs) in unlinked markers. Coding regions with high variability were identified by comparing whole genome sequences (WGS) from 43 human- and 92 ruminant-derived <em>C. parvum</em> samples collected across Europe. We first selected 18 markers and showed that they provide high discrimination among the samples with WGS data, with 88% of the MLSTs being singletons. Next, we defined a MLST based on eight genetically unlinked markers and generated sequence data from 305 <em>C. parvum</em> samples, collected from four different host species and 13 European countries. We consolidated a set of 365 fully genotyped samples, characterized by the presence of 154 different MLSTs, 105 of which were singletons. Network analyses showed no complete clustering of samples by host species or country of origin at the European scale. We further showed that samples with <em>gp60</em> subtypes that are common in Europe are divided into many MLSTs by the new scheme, highlighting its increased discriminatory ability. However, the applicability of the scheme in public health settings is limited by its cost, turnaround time, and scalability. To achieve discrimination of <em>C. parvum</em> samples based on SNPs, a large number of loci needs to be analysed, and this is feasible using amplicon sequencing technologies.</div></div>","PeriodicalId":94311,"journal":{"name":"Current research in parasitology & vector-borne diseases","volume":"8 ","pages":"Article 100308"},"PeriodicalIF":1.7000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design, development, and testing of a new multi-locus sequence typing scheme for the zoonotic pathogen Cryptosporidium parvum\",\"authors\":\"Karin Troell , Christen Rune Stensvold , Anna Rosa Sannella , Martha Betson , Emma Östlund , Rachel M. Chalmers , Umer Chaudhry , Rebecca Davidson , Lauren Davies , Ralf Ignatius , Anton de Jong , Gregory Karadjian , Karim Adjou , Christian Klotz , Sokratis Ptochos , Guy Robinson , Jeroen Roelfsema , Barbara Soba , Jacek Sroka , Paolo Vatta , Simone M. Cacciò\",\"doi\":\"10.1016/j.crpvbd.2025.100308\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>The zoonotic parasite <em>Cryptosporidium parvum</em> is an important global cause of diarrheal disease in humans and young ruminants. Molecular typing is essential to track transmission routes and identify clusters of cases. Here, we developed a novel multi-locus sequence typing (MLST) scheme based on single nucleotide polymorphisms (SNPs) in unlinked markers. Coding regions with high variability were identified by comparing whole genome sequences (WGS) from 43 human- and 92 ruminant-derived <em>C. parvum</em> samples collected across Europe. We first selected 18 markers and showed that they provide high discrimination among the samples with WGS data, with 88% of the MLSTs being singletons. Next, we defined a MLST based on eight genetically unlinked markers and generated sequence data from 305 <em>C. parvum</em> samples, collected from four different host species and 13 European countries. We consolidated a set of 365 fully genotyped samples, characterized by the presence of 154 different MLSTs, 105 of which were singletons. Network analyses showed no complete clustering of samples by host species or country of origin at the European scale. We further showed that samples with <em>gp60</em> subtypes that are common in Europe are divided into many MLSTs by the new scheme, highlighting its increased discriminatory ability. However, the applicability of the scheme in public health settings is limited by its cost, turnaround time, and scalability. To achieve discrimination of <em>C. parvum</em> samples based on SNPs, a large number of loci needs to be analysed, and this is feasible using amplicon sequencing technologies.</div></div>\",\"PeriodicalId\":94311,\"journal\":{\"name\":\"Current research in parasitology & vector-borne diseases\",\"volume\":\"8 \",\"pages\":\"Article 100308\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current research in parasitology & vector-borne diseases\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2667114X25000688\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PARASITOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in parasitology & vector-borne diseases","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2667114X25000688","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PARASITOLOGY","Score":null,"Total":0}
Design, development, and testing of a new multi-locus sequence typing scheme for the zoonotic pathogen Cryptosporidium parvum
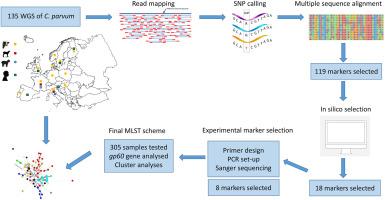
The zoonotic parasite Cryptosporidium parvum is an important global cause of diarrheal disease in humans and young ruminants. Molecular typing is essential to track transmission routes and identify clusters of cases. Here, we developed a novel multi-locus sequence typing (MLST) scheme based on single nucleotide polymorphisms (SNPs) in unlinked markers. Coding regions with high variability were identified by comparing whole genome sequences (WGS) from 43 human- and 92 ruminant-derived C. parvum samples collected across Europe. We first selected 18 markers and showed that they provide high discrimination among the samples with WGS data, with 88% of the MLSTs being singletons. Next, we defined a MLST based on eight genetically unlinked markers and generated sequence data from 305 C. parvum samples, collected from four different host species and 13 European countries. We consolidated a set of 365 fully genotyped samples, characterized by the presence of 154 different MLSTs, 105 of which were singletons. Network analyses showed no complete clustering of samples by host species or country of origin at the European scale. We further showed that samples with gp60 subtypes that are common in Europe are divided into many MLSTs by the new scheme, highlighting its increased discriminatory ability. However, the applicability of the scheme in public health settings is limited by its cost, turnaround time, and scalability. To achieve discrimination of C. parvum samples based on SNPs, a large number of loci needs to be analysed, and this is feasible using amplicon sequencing technologies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


