Yeong Jin Ahn, Yun Ho Lee, Su Hyun Park, Jeong Wook Kim, Cheul Hyun Yoon, Seongjun Kim, Young Jae Park and Byoung Don Kong*,
{"title":"用界面缺陷动力学模拟SiC氧化和退火的反应键序势","authors":"Yeong Jin Ahn, Yun Ho Lee, Su Hyun Park, Jeong Wook Kim, Cheul Hyun Yoon, Seongjun Kim, Young Jae Park and Byoung Don Kong*, ","doi":"10.1021/acs.jpcc.5c03173","DOIUrl":null,"url":null,"abstract":"<p >A newly developed Tersoff-type bond-order potential is presented for molecular dynamics (MD) simulations of silicon carbide (SiC) oxidation and postoxidation annealing processes. The potential encompasses interactions among Si, C, O, N, and H atoms and was parametrized using density functional theory (DFT) calculations, with experimental calibration to ensure predictive accuracy. The potential reliably reproduces key structural and thermodynamic properties of SiC-related compounds, as validated through MD simulations of oxidation and annealing. Simulations of thermal oxidation demonstrated the formation of a SiO<sub>2</sub> layer with density and Si–O bond lengths consistent with theoretical expectations, along with the emergence of interfacial carbon-related defects. N<sub>2</sub> annealing simulations revealed dynamic defect evolution, with increased N<sub>2</sub> pressure accelerating carbon removal and oxygen loss. Notably, elevated pressure conditions led to a significant reduction in the level of C–C bond formation, indicating the potential for defect mitigation through annealing control. Importantly, seven previously unreported defect types were identified at the SiC/SiO<sub>2</sub> interface, which were further classified into 11 unique atomic configurations and characterized using DFT. The simulated defect distribution exhibits strong correlation with experimentally observed interface trap densities, providing insight into fast and deep-level trap formation in SiC devices. This work establishes a robust atomistic modeling framework for realistic simulations of SiC oxidation and annealing and offers new avenues for interface defect engineering in next-generation SiC-based electronics.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 33","pages":"15029–15040"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Reactive Bond-Order Potential for Modeling SiC Oxidation and Annealing with Interface Defect Dynamics\",\"authors\":\"Yeong Jin Ahn, Yun Ho Lee, Su Hyun Park, Jeong Wook Kim, Cheul Hyun Yoon, Seongjun Kim, Young Jae Park and Byoung Don Kong*, \",\"doi\":\"10.1021/acs.jpcc.5c03173\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A newly developed Tersoff-type bond-order potential is presented for molecular dynamics (MD) simulations of silicon carbide (SiC) oxidation and postoxidation annealing processes. The potential encompasses interactions among Si, C, O, N, and H atoms and was parametrized using density functional theory (DFT) calculations, with experimental calibration to ensure predictive accuracy. The potential reliably reproduces key structural and thermodynamic properties of SiC-related compounds, as validated through MD simulations of oxidation and annealing. Simulations of thermal oxidation demonstrated the formation of a SiO<sub>2</sub> layer with density and Si–O bond lengths consistent with theoretical expectations, along with the emergence of interfacial carbon-related defects. N<sub>2</sub> annealing simulations revealed dynamic defect evolution, with increased N<sub>2</sub> pressure accelerating carbon removal and oxygen loss. Notably, elevated pressure conditions led to a significant reduction in the level of C–C bond formation, indicating the potential for defect mitigation through annealing control. Importantly, seven previously unreported defect types were identified at the SiC/SiO<sub>2</sub> interface, which were further classified into 11 unique atomic configurations and characterized using DFT. The simulated defect distribution exhibits strong correlation with experimentally observed interface trap densities, providing insight into fast and deep-level trap formation in SiC devices. This work establishes a robust atomistic modeling framework for realistic simulations of SiC oxidation and annealing and offers new avenues for interface defect engineering in next-generation SiC-based electronics.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 33\",\"pages\":\"15029–15040\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-08-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03173\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c03173","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Reactive Bond-Order Potential for Modeling SiC Oxidation and Annealing with Interface Defect Dynamics
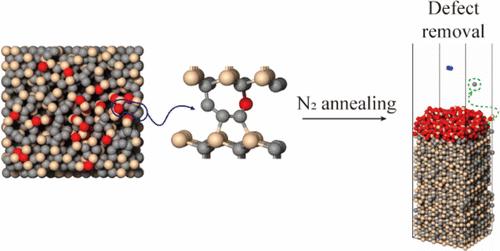
A newly developed Tersoff-type bond-order potential is presented for molecular dynamics (MD) simulations of silicon carbide (SiC) oxidation and postoxidation annealing processes. The potential encompasses interactions among Si, C, O, N, and H atoms and was parametrized using density functional theory (DFT) calculations, with experimental calibration to ensure predictive accuracy. The potential reliably reproduces key structural and thermodynamic properties of SiC-related compounds, as validated through MD simulations of oxidation and annealing. Simulations of thermal oxidation demonstrated the formation of a SiO2 layer with density and Si–O bond lengths consistent with theoretical expectations, along with the emergence of interfacial carbon-related defects. N2 annealing simulations revealed dynamic defect evolution, with increased N2 pressure accelerating carbon removal and oxygen loss. Notably, elevated pressure conditions led to a significant reduction in the level of C–C bond formation, indicating the potential for defect mitigation through annealing control. Importantly, seven previously unreported defect types were identified at the SiC/SiO2 interface, which were further classified into 11 unique atomic configurations and characterized using DFT. The simulated defect distribution exhibits strong correlation with experimentally observed interface trap densities, providing insight into fast and deep-level trap formation in SiC devices. This work establishes a robust atomistic modeling framework for realistic simulations of SiC oxidation and annealing and offers new avenues for interface defect engineering in next-generation SiC-based electronics.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


