Bénédicte L Tremblay, Anne-Marie Madore, Catherine Laprise
{"title":"法裔加拿大人食物致敏的DNA甲基化。","authors":"Bénédicte L Tremblay, Anne-Marie Madore, Catherine Laprise","doi":"10.1186/s13148-025-01951-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Food allergy (FA) is a great public health concern with an increased prevalence in the last decades. The underlying development mechanisms of FA and food sensitization (FS), which represents the first stage of development of FA, are influenced by environmental, epigenetic, and genetic factors. DNA methylation is an important epigenetic mediator of gene-environment interactions and key to understanding these mechanisms. Studies have linked whole-genome DNA methylation profile to FA and FS, but they all use methylation arrays. Methylation sequencing captures target regions of methylome with an extensive coverage. Thus, our objective was to identify CpG sites in genome-wide immune regulatory regions associated with FS and test their association with genetic variants using methylation quantitative trait loci (mQTL) analysis in French-Canadian individuals.</p><p><strong>Results: </strong>In 114 individuals from the Saguenay-Lac-Saint-Jean asthma family cohort, a total of 10 CpG sites out of 5,233,004 CpG sites were associated with the FS status (P < 1 × 10<sup>-8</sup>). CpG sites were located in 10 genes (ARRDC1, B9D1, CFL1, CROCC, DHX36, DND1, PMS1, RASSF1, TOP2A, and USP21) all of which were associated with immune response and allergic diseases (allergy, asthma, atopic dermatitis, and allergic rhinitis) in the literature. Almost all individuals with FS clustered based on the methylation levels of these CpG sites which reinforces their importance and relevance to discriminate individuals with and without FS in our cohort. Among the top 10 most significant CpG sites, five were associated with 478 genetic variants in trans-mQTL (FDR < 1 × 10<sup>-8</sup>).</p><p><strong>Conclusions: </strong>To our knowledge, this is a unique association study between FS and DNA methylation using targeted bisulfite sequencing across the genome. This approach provides high-resolution assessment of genome-wide functional methylome that yields valuable understandings to this field of research. The results reveal potential relationships between FS, CpG sites, and genetic variants located in genes involved in allergic diseases. This provides potential insights on the underlying effects of DNA methylation and genetic variants on FS and possibly the pathogenesis of FA. Further epigenome-wide studies on larger samples combined with genome-wide genotyping are needed to validate the results and verify the biological potential of these CpG sites.</p>","PeriodicalId":10366,"journal":{"name":"Clinical Epigenetics","volume":"17 1","pages":"143"},"PeriodicalIF":4.4000,"publicationDate":"2025-08-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12359973/pdf/","citationCount":"0","resultStr":"{\"title\":\"DNA methylation of food sensitization in a French-Canadian population.\",\"authors\":\"Bénédicte L Tremblay, Anne-Marie Madore, Catherine Laprise\",\"doi\":\"10.1186/s13148-025-01951-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Food allergy (FA) is a great public health concern with an increased prevalence in the last decades. The underlying development mechanisms of FA and food sensitization (FS), which represents the first stage of development of FA, are influenced by environmental, epigenetic, and genetic factors. DNA methylation is an important epigenetic mediator of gene-environment interactions and key to understanding these mechanisms. Studies have linked whole-genome DNA methylation profile to FA and FS, but they all use methylation arrays. Methylation sequencing captures target regions of methylome with an extensive coverage. Thus, our objective was to identify CpG sites in genome-wide immune regulatory regions associated with FS and test their association with genetic variants using methylation quantitative trait loci (mQTL) analysis in French-Canadian individuals.</p><p><strong>Results: </strong>In 114 individuals from the Saguenay-Lac-Saint-Jean asthma family cohort, a total of 10 CpG sites out of 5,233,004 CpG sites were associated with the FS status (P < 1 × 10<sup>-8</sup>). CpG sites were located in 10 genes (ARRDC1, B9D1, CFL1, CROCC, DHX36, DND1, PMS1, RASSF1, TOP2A, and USP21) all of which were associated with immune response and allergic diseases (allergy, asthma, atopic dermatitis, and allergic rhinitis) in the literature. Almost all individuals with FS clustered based on the methylation levels of these CpG sites which reinforces their importance and relevance to discriminate individuals with and without FS in our cohort. Among the top 10 most significant CpG sites, five were associated with 478 genetic variants in trans-mQTL (FDR < 1 × 10<sup>-8</sup>).</p><p><strong>Conclusions: </strong>To our knowledge, this is a unique association study between FS and DNA methylation using targeted bisulfite sequencing across the genome. This approach provides high-resolution assessment of genome-wide functional methylome that yields valuable understandings to this field of research. The results reveal potential relationships between FS, CpG sites, and genetic variants located in genes involved in allergic diseases. This provides potential insights on the underlying effects of DNA methylation and genetic variants on FS and possibly the pathogenesis of FA. Further epigenome-wide studies on larger samples combined with genome-wide genotyping are needed to validate the results and verify the biological potential of these CpG sites.</p>\",\"PeriodicalId\":10366,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"17 1\",\"pages\":\"143\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-08-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12359973/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-025-01951-8\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-025-01951-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:食物过敏(FA)是一个重大的公共卫生问题,在过去的几十年里越来越普遍。脂肪酸和食物致敏(FS)的潜在发育机制受到环境、表观遗传和遗传因素的影响,FS是脂肪酸发育的第一阶段。DNA甲基化是基因-环境相互作用的重要表观遗传介质,也是理解这些机制的关键。研究已经将全基因组DNA甲基化谱与FA和FS联系起来,但它们都使用甲基化阵列。甲基化测序以广泛的覆盖范围捕获甲基组的目标区域。因此,我们的目标是确定与FS相关的全基因组免疫调节区域的CpG位点,并使用甲基化数量性状位点(mQTL)分析法裔加拿大人个体,测试它们与遗传变异的关联。结果:在来自saguenay - la - saint - jean哮喘家族队列的114例个体中,5233,004个CpG位点中共有10个CpG位点与FS状态相关(P -8)。CpG位点位于10个基因(ARRDC1、B9D1、CFL1、CROCC、DHX36、DND1、PMS1、RASSF1、TOP2A和USP21)中,这些基因在文献中都与免疫应答和过敏性疾病(过敏、哮喘、特应性皮炎和变应性鼻炎)相关。几乎所有患有FS的个体都基于这些CpG位点的甲基化水平聚类,这加强了它们在我们的队列中区分患有和不患有FS的个体的重要性和相关性。在前10个最重要的CpG位点中,有5个位点与478个反式mqtl遗传变异(FDR -8)相关。结论:据我们所知,这是一个独特的FS和DNA甲基化之间的关联研究,使用靶向亚硫酸盐测序跨基因组。这种方法提供了全基因组功能甲基组的高分辨率评估,对这一研究领域产生了有价值的理解。结果揭示了FS、CpG位点和位于过敏性疾病相关基因的遗传变异之间的潜在关系。这为DNA甲基化和遗传变异对FS的潜在影响以及FA的可能发病机制提供了潜在的见解。需要在更大的样本上进行进一步的全基因组研究,并结合全基因组基因分型来验证结果并验证这些CpG位点的生物学潜力。
DNA methylation of food sensitization in a French-Canadian population.
Background: Food allergy (FA) is a great public health concern with an increased prevalence in the last decades. The underlying development mechanisms of FA and food sensitization (FS), which represents the first stage of development of FA, are influenced by environmental, epigenetic, and genetic factors. DNA methylation is an important epigenetic mediator of gene-environment interactions and key to understanding these mechanisms. Studies have linked whole-genome DNA methylation profile to FA and FS, but they all use methylation arrays. Methylation sequencing captures target regions of methylome with an extensive coverage. Thus, our objective was to identify CpG sites in genome-wide immune regulatory regions associated with FS and test their association with genetic variants using methylation quantitative trait loci (mQTL) analysis in French-Canadian individuals.
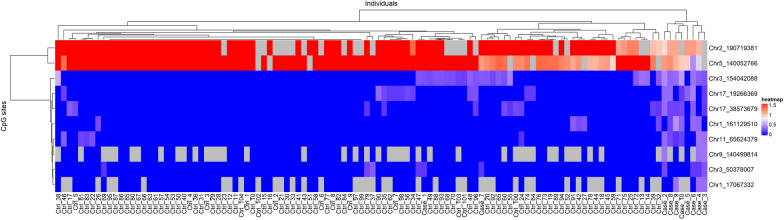
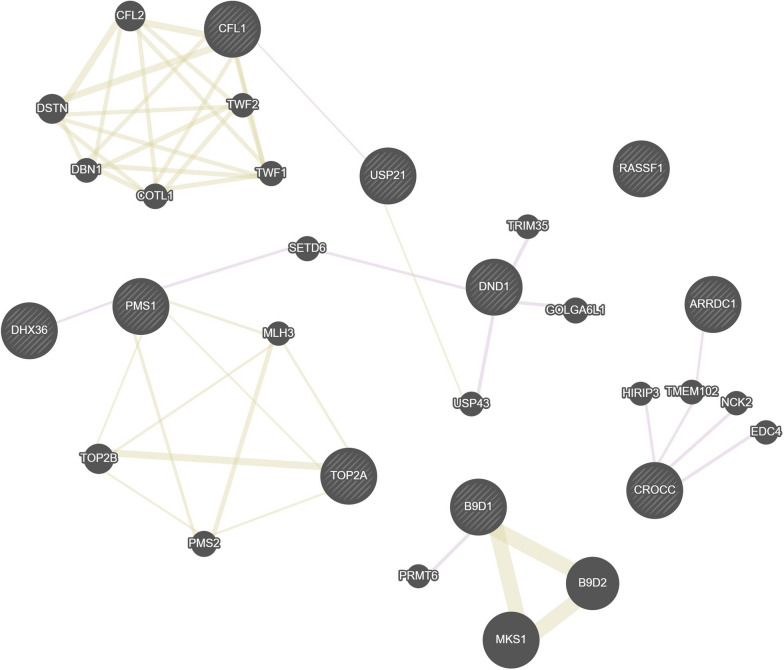
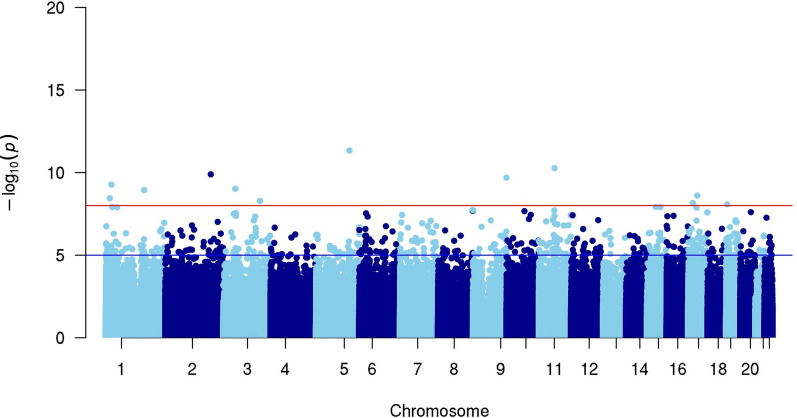
Results: In 114 individuals from the Saguenay-Lac-Saint-Jean asthma family cohort, a total of 10 CpG sites out of 5,233,004 CpG sites were associated with the FS status (P < 1 × 10-8). CpG sites were located in 10 genes (ARRDC1, B9D1, CFL1, CROCC, DHX36, DND1, PMS1, RASSF1, TOP2A, and USP21) all of which were associated with immune response and allergic diseases (allergy, asthma, atopic dermatitis, and allergic rhinitis) in the literature. Almost all individuals with FS clustered based on the methylation levels of these CpG sites which reinforces their importance and relevance to discriminate individuals with and without FS in our cohort. Among the top 10 most significant CpG sites, five were associated with 478 genetic variants in trans-mQTL (FDR < 1 × 10-8).
Conclusions: To our knowledge, this is a unique association study between FS and DNA methylation using targeted bisulfite sequencing across the genome. This approach provides high-resolution assessment of genome-wide functional methylome that yields valuable understandings to this field of research. The results reveal potential relationships between FS, CpG sites, and genetic variants located in genes involved in allergic diseases. This provides potential insights on the underlying effects of DNA methylation and genetic variants on FS and possibly the pathogenesis of FA. Further epigenome-wide studies on larger samples combined with genome-wide genotyping are needed to validate the results and verify the biological potential of these CpG sites.
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


