Abdul Wasim*, Lars V. Schäfer* and Jagannath Mondal*,
{"title":"应用人工神经网络实现分子动力学模拟轨迹的优化存储和便捷共享。","authors":"Abdul Wasim*, Lars V. Schäfer* and Jagannath Mondal*, ","doi":"10.1021/acs.jcim.5c01294","DOIUrl":null,"url":null,"abstract":"<p >With the remarkable stride in computing power and advances in Molecular Dynamics (MD) simulation programs, the crucial challenge of storing and sharing large biomolecular simulation data sets has emerged. By leveraging AutoEncoders, a type of artificial neural network, we developed a method to compress MD trajectories into significantly smaller latent spaces. Our method can save up to 98% in disk space compared to <span>xtc</span>, a highly compressed trajectory format from the widely used MD program package GROMACS, thus facilitating storage and sharing of simulation trajectories. Atom coordinates are very accurately reconstructed from compressed data. The method was tested across a diverse sets of biomolecular systems, including folded proteins, intrinsically disordered proteins, phospholipid bilayers, protein–ligand complexes, large protein complexes and membrane-bound protein systems. The reconstructed trajectories demonstrated consistent accuracy in recovering key biophysically relevant properties for proteins, lipids and composite systems. The compression efficiency was particularly beneficial for larger systems. This approach enables the scientific community to efficiently store and share large-scale biomolecular simulation data, potentially enhancing collaborative research efforts. The workflow, termed “compresstraj”, is implemented in PyTorch and is publicly available at https://github.com/SerpentByte/compresstraj, offering a practical solution for handling the increasing volumes of data generated in biomolecular simulation studies.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 17","pages":"9022–9033"},"PeriodicalIF":5.3000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Employing Artificial Neural Networks for Optimal Storage and Facile Sharing of Molecular Dynamics Simulation Trajectories\",\"authors\":\"Abdul Wasim*, Lars V. Schäfer* and Jagannath Mondal*, \",\"doi\":\"10.1021/acs.jcim.5c01294\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >With the remarkable stride in computing power and advances in Molecular Dynamics (MD) simulation programs, the crucial challenge of storing and sharing large biomolecular simulation data sets has emerged. By leveraging AutoEncoders, a type of artificial neural network, we developed a method to compress MD trajectories into significantly smaller latent spaces. Our method can save up to 98% in disk space compared to <span>xtc</span>, a highly compressed trajectory format from the widely used MD program package GROMACS, thus facilitating storage and sharing of simulation trajectories. Atom coordinates are very accurately reconstructed from compressed data. The method was tested across a diverse sets of biomolecular systems, including folded proteins, intrinsically disordered proteins, phospholipid bilayers, protein–ligand complexes, large protein complexes and membrane-bound protein systems. The reconstructed trajectories demonstrated consistent accuracy in recovering key biophysically relevant properties for proteins, lipids and composite systems. The compression efficiency was particularly beneficial for larger systems. This approach enables the scientific community to efficiently store and share large-scale biomolecular simulation data, potentially enhancing collaborative research efforts. The workflow, termed “compresstraj”, is implemented in PyTorch and is publicly available at https://github.com/SerpentByte/compresstraj, offering a practical solution for handling the increasing volumes of data generated in biomolecular simulation studies.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 17\",\"pages\":\"9022–9033\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c01294\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c01294","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
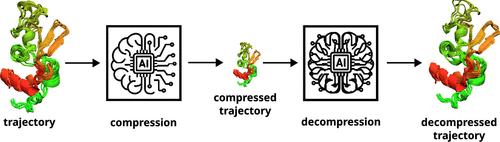
Employing Artificial Neural Networks for Optimal Storage and Facile Sharing of Molecular Dynamics Simulation Trajectories
With the remarkable stride in computing power and advances in Molecular Dynamics (MD) simulation programs, the crucial challenge of storing and sharing large biomolecular simulation data sets has emerged. By leveraging AutoEncoders, a type of artificial neural network, we developed a method to compress MD trajectories into significantly smaller latent spaces. Our method can save up to 98% in disk space compared to xtc, a highly compressed trajectory format from the widely used MD program package GROMACS, thus facilitating storage and sharing of simulation trajectories. Atom coordinates are very accurately reconstructed from compressed data. The method was tested across a diverse sets of biomolecular systems, including folded proteins, intrinsically disordered proteins, phospholipid bilayers, protein–ligand complexes, large protein complexes and membrane-bound protein systems. The reconstructed trajectories demonstrated consistent accuracy in recovering key biophysically relevant properties for proteins, lipids and composite systems. The compression efficiency was particularly beneficial for larger systems. This approach enables the scientific community to efficiently store and share large-scale biomolecular simulation data, potentially enhancing collaborative research efforts. The workflow, termed “compresstraj”, is implemented in PyTorch and is publicly available at https://github.com/SerpentByte/compresstraj, offering a practical solution for handling the increasing volumes of data generated in biomolecular simulation studies.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


