利用Se和Ga浓度调节Co2MnGe合金的结构、磁性、电子和力学特性:蒙特卡罗和DFT方法
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
采用蒙特卡罗模拟和第一性原理计算,研究了Co2MnGe1-xYx (Y= Ga和Se, x = 0,0.25, 0.50和0.75)化合物的结构、电子、磁性和光学特性。这些化合物的计算结构特征表明,当Se和Ga取代x增加时,晶格常数也略有增加。在其最佳晶格常数下,带结构计算预测这些化合物是半金属铁磁体,用于Se取代,半金属铁磁体用于Ga取代。当Ge原子部分被Se和Ga取代时,原子磁矩和总磁矩保持不变。计算得到的化合物的磁矩值与理论和实验结果吻合得很好。大的自旋极化和半金属性是自旋电子学应用所需要的。在H = 4 T时,比热和磁熵变化达到最大值,分别为75.30 J mol−1 K−1和0.72 J kg−1 K−1。本文章由计算机程序翻译,如有差异,请以英文原文为准。
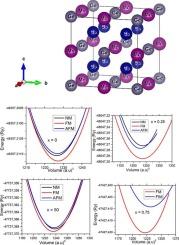
Using Se and Ga concentration to adjust the structural, magnetic, electronic and mechanical characteristics of Co2MnGe alloy: A Monte Carlo and DFT approach
Co2MnGe1-xYx (Y= Ga and Se with x = 0, 0.25, 0.50 and 0.75) compounds structural, electronic, magnetic, and optical characteristics are examined using Monte Carlo simulation and first-principles calculations using the generalized gradient approximation. These compounds computed structural characteristics indicate that when Se and Ga substitution x increase, the lattice constants marginally increase as well. At their optimal lattice constants, the band structure computations predict that these compounds are half-metallic ferrimagnets for Se substitution and half-metallic ferromagnets for Ga substitution. The atomic and total magnetic moments remain unchanged when the Ge atom is partially replaced by Se and Ga. The calculated magnetic moment values of our compounds accord well with both theoretical and experimental findings. Large spin polarization and half-metallicity are desired for applications in spintronics. At H = 4 T, the specific heat and magnetic entropy changes reach their maximums of 75.30 J mol−1 K−1 and 0.72 J kg−1 K−1.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


