利用整合序列和基于人工智能的结构方法对脓肿分枝杆菌蛋白质组进行功能(重新)注释
IF 2.7
Q3 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
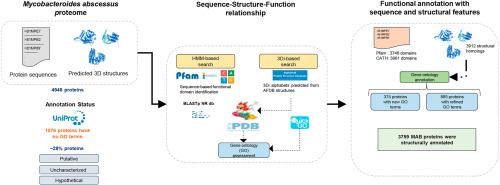
蛋白质的功能注释对于理解生物体的基本生物学至关重要。在病原体的背景下,它可以为其功能景观提供有价值的见解,有助于理解发病和生存的分子机制。在这项研究中,我们探索了序列和人工智能驱动的基于结构的方法对脓肿分枝杆菌(Mycobacteroides abessus, MAB)进行功能性(重新)注释的应用。单克隆抗体是一种机会性病原体,可引起免疫功能低下患者的感染,并对几种抗生素具有耐药性。全球耐药菌株的增加以及最近确定的间接人际传播的可能性强调了将单克隆抗体理解为一种关键病原体的重要性。然而,我们对单克隆抗体蛋白质组的理解存在巨大差距,这不仅对了解各种蛋白质的功能方面至关重要,而且对于优先考虑治疗开发的药物靶点也至关重要。目前,在UniProtKB中,28%的MAB蛋白质组的注释很差,超过四分之一的MAB蛋白质组缺乏基因本体(GO)术语,表明缺乏标准的功能描述。为此,本研究旨在使用基于序列和结构的方法相结合,以系统的方式对MAB蛋白质组进行功能(重新)注释。我们对NR数据库进行了基于序列的相似性搜索,并使用Pfam和CATH进行了基于HMM的功能域搜索。然后,我们利用MAB alphafold预测结构对MAB蛋白进行基于结构的相似性搜索,使用Foldseek来识别蛋白质并转移其基因本体(GO)注释。我们分配了新的GO注释(374个蛋白质),并改进了先前未注释的MAB必需基因的现有注释(885个蛋白质)。此外,我们还使用基于综合序列和结构的方法对29个无法获得AlphaFold结构的蛋白质进行了注释。最后,研究人员对一些与结核分枝杆菌相似的蛋白进行了结构比较,揭示了MAB中与耐药性相关的残留水平差异。我们的研究强调了一种结合序列和人工智能驱动的基于结构的大规模蛋白质组功能注释方法,该方法可应用于任何感兴趣的生物体。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Functional (re)annotation of Mycobacteroides abscessus proteome using integrative sequence and AI-based structural approaches
Functional annotation of proteins is crucial in understanding the basic biology of organisms. In the context of pathogens, it can provide valuable insights towards its functional landscape contributing to understanding the molecular mechanisms of pathogenesis and survival. In this study, we explored the applications of sequence and AI-driven structure-based methods to functionally (re)annotate Mycobacteroides abscessus (MAB). MAB is an opportunistic pathogen responsible for causing infections in immunocompromised patients and exhibits resistance to several antibiotics. The global rise in drug-resistant strains and the recently identified potential for indirect human-to-human transmission emphasizes the importance of understanding MAB as a critical pathogen. However, there is a huge gap in our understanding of the MAB proteome, which is vital not only for understanding the functional aspects of various proteins but also for prioritizing drug targets for therapeutic development. Presently, 28 % of the MAB proteome, as available in UniProtKB, is poorly annotated, and more than a fourth of MAB proteome lack gene ontology (GO) terms, indicating a lack of standard functional descriptions. To this end, the present study aims to functionally (re)annotate MAB proteome using a combination of sequence and structure-based approaches in a systematic way. We performed sequence-based similarity search against NR database and performed HMM based search for functional domains with Pfam and CATH. Then, we utilized MAB AlphaFold-predicted structures to annotate MAB proteins with structure-based similarity search using Foldseek to identify proteins and transfer their gene ontology (GO) annotations. We assigned new GO annotations (374 proteins) and refined the existing annotations (885 proteins) for previously unannotated essential genes of MAB. In addition, we also performed annotations using an integrated sequence and structure-based approach for the 29 proteins for which AlphaFold structures were not available. In the end, structural comparisons of a few proteins that were similar to Mycobacterium tuberculosis were explored, revealing residue-level differences in MAB linked to drug resistance. Our study highlights a combined sequence- and AI-driven structure-based approach for large-scale proteome functional annotation, which can be applied to any organism of interest.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


