Artem Shagurin, Michael G. Kiselev, Pal Jedlovszky, Natalia T. Correia, Frederic Affouard and Abdenacer Idrissi*,
{"title":"姜黄素多晶体相的热行为和局部结构组织:分子动力学模拟分析。","authors":"Artem Shagurin, Michael G. Kiselev, Pal Jedlovszky, Natalia T. Correia, Frederic Affouard and Abdenacer Idrissi*, ","doi":"10.1021/acs.jpcb.5c04240","DOIUrl":null,"url":null,"abstract":"<p >Curcumin (CUR), a bioactive compound with known polymorphism, exhibits distinct conformational and thermophysical properties across its three crystalline forms. In this study, we employ molecular dynamics simulations to investigate the thermal behavior, local structural organization, and polymorph-specific stability of CUR in the bulk phase. We first evaluate four widely used classical force fields (OPLS-AA, CGENFF, GAFF2, and GROMOS) against experimental melting points, densities, and conformational preferences, identifying OPLS-AA as the most suitable one. Through targeted reparametrization of intramolecular dihedral angles based on DFT benchmarks, we significantly improve the ability of this force field to reproduce conformational distributions and melting transitions. Using the refined model, we characterize the temperature dependence of several structural observables, including local density (via Voronoi tessellation), nearest-neighbor distributions, pair interaction energies, hydrogen bonding, and molecular orientation. Our results reveal that conformational transitions, packing rearrangements, and fluctuations occur cooperatively near polymorph-specific temperatures, often beginning with disrupted π–π stacking and propagating through the lattice. Notably, cooling simulations fail to induce recrystallization, resulting in amorphous states. This comprehensive analysis highlights the critical interplay between molecular conformation, packing, and directional interactions in determining CUR’s polymorphic behavior, providing a mechanistic foundation for controlling phase transitions in flexible molecular solids.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"129 33","pages":"8615–8625"},"PeriodicalIF":2.9000,"publicationDate":"2025-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Thermal Behavior and Local Structural Organization in Curcumin Polymorphs’ Bulk Phase: A Molecular Dynamics Simulation Analysis\",\"authors\":\"Artem Shagurin, Michael G. Kiselev, Pal Jedlovszky, Natalia T. Correia, Frederic Affouard and Abdenacer Idrissi*, \",\"doi\":\"10.1021/acs.jpcb.5c04240\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Curcumin (CUR), a bioactive compound with known polymorphism, exhibits distinct conformational and thermophysical properties across its three crystalline forms. In this study, we employ molecular dynamics simulations to investigate the thermal behavior, local structural organization, and polymorph-specific stability of CUR in the bulk phase. We first evaluate four widely used classical force fields (OPLS-AA, CGENFF, GAFF2, and GROMOS) against experimental melting points, densities, and conformational preferences, identifying OPLS-AA as the most suitable one. Through targeted reparametrization of intramolecular dihedral angles based on DFT benchmarks, we significantly improve the ability of this force field to reproduce conformational distributions and melting transitions. Using the refined model, we characterize the temperature dependence of several structural observables, including local density (via Voronoi tessellation), nearest-neighbor distributions, pair interaction energies, hydrogen bonding, and molecular orientation. Our results reveal that conformational transitions, packing rearrangements, and fluctuations occur cooperatively near polymorph-specific temperatures, often beginning with disrupted π–π stacking and propagating through the lattice. Notably, cooling simulations fail to induce recrystallization, resulting in amorphous states. This comprehensive analysis highlights the critical interplay between molecular conformation, packing, and directional interactions in determining CUR’s polymorphic behavior, providing a mechanistic foundation for controlling phase transitions in flexible molecular solids.</p>\",\"PeriodicalId\":60,\"journal\":{\"name\":\"The Journal of Physical Chemistry B\",\"volume\":\"129 33\",\"pages\":\"8615–8625\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-08-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry B\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c04240\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.5c04240","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
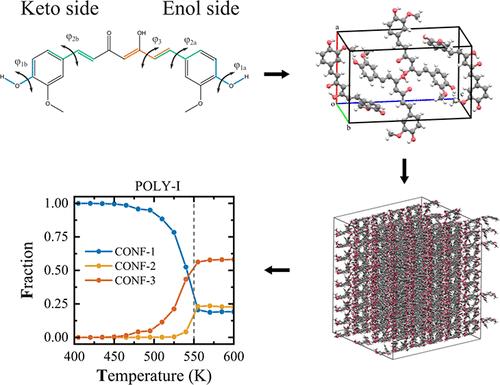
Thermal Behavior and Local Structural Organization in Curcumin Polymorphs’ Bulk Phase: A Molecular Dynamics Simulation Analysis
Curcumin (CUR), a bioactive compound with known polymorphism, exhibits distinct conformational and thermophysical properties across its three crystalline forms. In this study, we employ molecular dynamics simulations to investigate the thermal behavior, local structural organization, and polymorph-specific stability of CUR in the bulk phase. We first evaluate four widely used classical force fields (OPLS-AA, CGENFF, GAFF2, and GROMOS) against experimental melting points, densities, and conformational preferences, identifying OPLS-AA as the most suitable one. Through targeted reparametrization of intramolecular dihedral angles based on DFT benchmarks, we significantly improve the ability of this force field to reproduce conformational distributions and melting transitions. Using the refined model, we characterize the temperature dependence of several structural observables, including local density (via Voronoi tessellation), nearest-neighbor distributions, pair interaction energies, hydrogen bonding, and molecular orientation. Our results reveal that conformational transitions, packing rearrangements, and fluctuations occur cooperatively near polymorph-specific temperatures, often beginning with disrupted π–π stacking and propagating through the lattice. Notably, cooling simulations fail to induce recrystallization, resulting in amorphous states. This comprehensive analysis highlights the critical interplay between molecular conformation, packing, and directional interactions in determining CUR’s polymorphic behavior, providing a mechanistic foundation for controlling phase transitions in flexible molecular solids.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


