{"title":"共价抑制的动力学模型:快速波动中间态的影响。","authors":"Kyle Ghaby, and , Benoît Roux*, ","doi":"10.1021/acs.jcim.5c00952","DOIUrl":null,"url":null,"abstract":"<p >There is increasing interest in the discovery of small-molecule inhibitors that form covalent bonds with their targets for therapeutic applications. Nevertheless, identifying clear rational design principles remains challenging because the action of these molecules cannot be understood as common noncovalent inhibition. Conventional kinetic models often reduce the binding of covalent inhibitors to a two-step irreversible process, overlooking rapid complex dynamics of the associated unlinked inhibitor before the formation of the covalent bond with its target. In the present analysis, we expand the intermediate state into two conformations: reactive (E·I) and nonreactive (E··I). To illustrate the consequences of such simplification, the expanded kinetic model can be reduced to an effective two-step scheme expressed in terms of the equilibrium probability of the unlinked inhibitor to form either conformation. A mass-action-based numerical workflow is implemented to simulate time-dependent kinetics, overcoming the common limitations of empirical models. The numerical workflow helps relate microscopic states observed in molecular dynamics simulations to macroscopic observables like EC<sub>50</sub> and the apparent rate of covalent inhibition, showing the impact of transient intermediates on dissociation rates and potency. The proposed framework refines the interpretation of dose-response data, aiding medicinal chemists in optimizing covalent inhibitors and providing a quantitative platform that relates molecular conformational distributions to empirical parameters.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 16","pages":"8730–8739"},"PeriodicalIF":5.3000,"publicationDate":"2025-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Kinetic Modeling of Covalent Inhibition: Effects of Rapidly Fluctuating Intermediate States\",\"authors\":\"Kyle Ghaby, and , Benoît Roux*, \",\"doi\":\"10.1021/acs.jcim.5c00952\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >There is increasing interest in the discovery of small-molecule inhibitors that form covalent bonds with their targets for therapeutic applications. Nevertheless, identifying clear rational design principles remains challenging because the action of these molecules cannot be understood as common noncovalent inhibition. Conventional kinetic models often reduce the binding of covalent inhibitors to a two-step irreversible process, overlooking rapid complex dynamics of the associated unlinked inhibitor before the formation of the covalent bond with its target. In the present analysis, we expand the intermediate state into two conformations: reactive (E·I) and nonreactive (E··I). To illustrate the consequences of such simplification, the expanded kinetic model can be reduced to an effective two-step scheme expressed in terms of the equilibrium probability of the unlinked inhibitor to form either conformation. A mass-action-based numerical workflow is implemented to simulate time-dependent kinetics, overcoming the common limitations of empirical models. The numerical workflow helps relate microscopic states observed in molecular dynamics simulations to macroscopic observables like EC<sub>50</sub> and the apparent rate of covalent inhibition, showing the impact of transient intermediates on dissociation rates and potency. The proposed framework refines the interpretation of dose-response data, aiding medicinal chemists in optimizing covalent inhibitors and providing a quantitative platform that relates molecular conformational distributions to empirical parameters.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 16\",\"pages\":\"8730–8739\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00952\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00952","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Kinetic Modeling of Covalent Inhibition: Effects of Rapidly Fluctuating Intermediate States
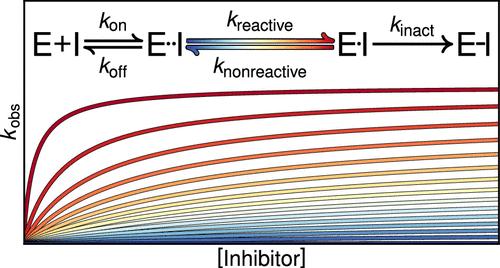
There is increasing interest in the discovery of small-molecule inhibitors that form covalent bonds with their targets for therapeutic applications. Nevertheless, identifying clear rational design principles remains challenging because the action of these molecules cannot be understood as common noncovalent inhibition. Conventional kinetic models often reduce the binding of covalent inhibitors to a two-step irreversible process, overlooking rapid complex dynamics of the associated unlinked inhibitor before the formation of the covalent bond with its target. In the present analysis, we expand the intermediate state into two conformations: reactive (E·I) and nonreactive (E··I). To illustrate the consequences of such simplification, the expanded kinetic model can be reduced to an effective two-step scheme expressed in terms of the equilibrium probability of the unlinked inhibitor to form either conformation. A mass-action-based numerical workflow is implemented to simulate time-dependent kinetics, overcoming the common limitations of empirical models. The numerical workflow helps relate microscopic states observed in molecular dynamics simulations to macroscopic observables like EC50 and the apparent rate of covalent inhibition, showing the impact of transient intermediates on dissociation rates and potency. The proposed framework refines the interpretation of dose-response data, aiding medicinal chemists in optimizing covalent inhibitors and providing a quantitative platform that relates molecular conformational distributions to empirical parameters.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


